Authors: Gary Welz
CopernicusAI, New York, NY; Person of Interest, CUNY Graduate Center
Abstract
We apply topological data analysis (TDA) to 108 genetic regulatory circuits from the Genome Logic Modeling Project (GLMP). Each circuit is represented as a Mermaid Markdown flowchart from which we extract five features: nodes, conditionals (edges), OR gates, AND gates, and loops (back-edges); flowcharts are generated with LLM assistance, making it feasible to produce and curate many such diagrams from a single prompt in seconds—a methodological breakthrough relative to earlier, labor-intensive manual charting. We encode each flowchart as a 5-dimensional feature vector and compute persistent homology (Vietoris–Rips, Ripser, maxdim=2). The most persistent H₁ loops align with known feedback and stress-response circuits: the top loop (persistence 0.563) aggregates SOS response, quorum sensing, biofilm formation, and protein quality control; a second loop (0.443) links antibiotic efflux, arginine and tryptophan biosynthesis, and stress response; ara operon and Pho regulon appear in Loop #5 (0.198). Topology groups processes by regulatory logic (e.g. negative feedback, stress response) rather than by pathway alone, including across organisms (E. coli, S. cerevisiae, Bacillus subtilis). We conclude that TDA on structural features captures genuine regulatory architecture and discuss limitations, biological coherence checks, and next steps: Mapper, persistent cohomology, and scaling to hundreds of processes with domain-expert validation.
Keywords: persistent homology, genetic circuits, feedback loops, regulatory networks, topological data analysis, Mermaid visualization, LLM-assisted curation
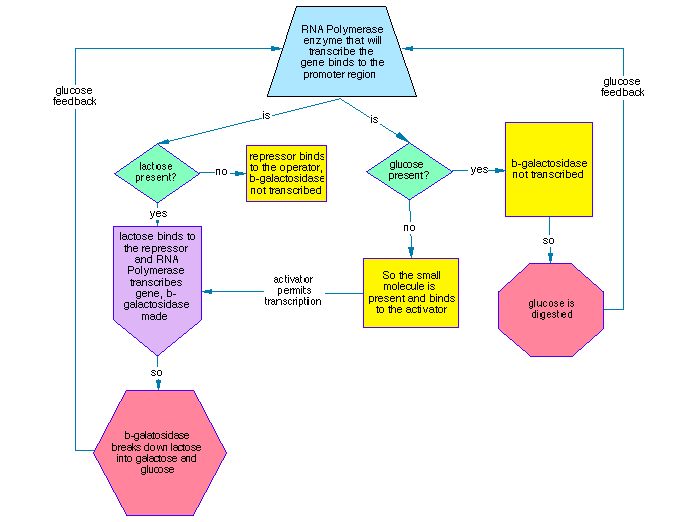
The idea of representing genetic regulatory processes as flowcharts has a long history. A first attempt at a β-galactosidase / Lac operon flowchart appeared in 1995 in an article in The X Advisor, an online magazine for Unix developers, entitled “Is the Genome Like a Computer Program?”, drawing on conversations with biologists on the bionet.genome.chromosome newsgroup. The original thread is archived at bio.net: first posting (April 1995), in which the genome was proposed as a flowchart with genes connected by logical “and” and “or”; replies from Robert Robbins (000539, 000543) and G. Dellaire (000540) raised points that remain pertinent. Robbins noted that “flow charts describe the behavior of a non-parallel machine” and that “care must be taken in interpreting that flow chart,” while also allowing that “bringing computer-science insights to bear on the challenge of understanding genome operation has some potentially huge payoffs.” Dellaire emphasized that “the actual structure of genome and not just the linear sequence may ‘encode’ sets of instructions for the ‘reading and accessing’ of this genetic code,” with context “spatial, what tissue; or temporal, what time of development”—a second level of language beyond the linear code. That distinction (logical/structure vs. sequence, and context) motivates the present use of flowcharts as structural data and the longer-term goal of linking topology to sequence motifs. The X Advisor article is archived at the Internet Archive; the newsgroup is also accessible via Google Groups. The β-galactosidase description used for that chart came from Berg & Singer (1992, pp. 71–73). Notably, the 1995 chart was created from text alone—the same process that LLMs use today: words describing a process → diagram. This methodological continuity shows that diagrams are only as detailed and reliable as their source material; using different sources for the same process can yield different charts, which explains why validation and fact-checking are essential. Producing such charts by hand was so time-consuming that the approach lay dormant for decades.
Figure 1. First β-galactosidase / Lac operon flowchart (1995). The chart was created from the textual description in Berg & Singer (1992, pp. 71–73) for the article “Is the Genome Like a Computer Program?” in The X Advisor; the article is archived at the Internet Archive. Produced by hand from text alone—the same process LLMs use today—it illustrates promoter–operator–repressor logic and lactose induction of β-galactosidase.
Recent advances in large language models (LLMs) and the adoption of Mermaid Markdown as a standard for structured diagrams have changed the picture: we can now generate and refine flowcharts from textual descriptions (e.g. paper excerpts) in seconds from a single prompt. The same Lac operon / β-galactosidase idea can be realized today as a Mermaid flowchart in the Genome Logic Modeling Project (GLMP) viewer, alongside 107 other genetic regulatory circuits. That methodological breakthrough—text to visual data at scale—motivates the present work.
We ask whether the shape of these circuits, as captured by topology, aligns with what biologists already know: feedback loops, cascades, and regulatory motifs. Feedback loops are literally loops; they should appear as persistent H₁ features. Can text-derived visual data support that?
Traditional TDA pipeline: Numerical measurements → feature vectors → topology.
This work: Text (e.g. papers) → Mermaid flowcharts → feature extraction → topology.
The key innovation is treating flowcharts as visual data objects. Mermaid Markdown converts textual process descriptions into structured diagrams with nodes, edges, and explicit OR/AND logic. We do not use the full graph for TDA; we summarize each flowchart into five numerical features (nodes, conditionals, OR gates, AND gates, loops). Those features are then used to build a distance between processes and to compute persistent homology. Thus we extract topology from descriptions (via their visual representation), not from direct numerical measurements. That shift opens the possibility of analyzing processes for which quantitative data are scarce or incomplete.
Conceptually, our approach resembles the Politics case study in Carlsson & Vejdemo-Johansson (2021, pp. 199–201): discrete entities (here, circuits) with derived features, analyzed via persistent homology to reveal structural groupings. Our application, however, exhibits several characteristics that distinguish it from other known TDA applications: (1) a text-to-visual-to-topology pipeline—we start from textual process descriptions, not numerical measurements or pre-existing point clouds; (2) five structural features (nodes, conditionals, OR gates, AND gates, loops) capturing circuit complexity and feedback, enabling a “programmable” interpretation of regulatory logic; (3) biological interpretability—feedback loops in biology correspond literally to H₁ loops in homology; and (4) LLM-assisted curation at scale, allowing rapid generation and refinement of flowcharts from a single prompt.
The Genome Logic Modeling Project (GLMP) provides 108 genetic
regulatory circuits: - E. coli: 66 processes
- S. cerevisiae: 38 processes
- Bacillus subtilis: 4 processes
Each process is represented as a Mermaid Markdown flowchart with nodes (genes, proteins, metabolites, conditions), edges (activation, repression, synthesis, degradation), and logic gates (OR, AND). We extract five features per process for TDA. Two-component signaling (EnvZ–OmpR in E. coli) is the subject of ongoing structural and mechanistic investigation (Rivera-Cancel et al., 2014; Swingle et al., 2025), and its representation in GLMP provides a concrete link between topological structure and regulatory logic. Examples include lac operon, SOS response, two-component EnvZ–OmpR signaling, ara and trp operons, heat shock, catabolite repression, and Pho regulon.
Each process JSON includes references (PubMed, DOI) so flowcharts are citable; the GLMP viewer accepts community feedback so diagrams are correctable. Code and data: https://github.com/garywelz/glmp. Interactive table and viewer: https://storage.googleapis.com/regal-scholar-453620-r7-podcast-storage/glmp-database-table.html.
We do not use the full graph structure for TDA. From each Mermaid flowchart we extract five numerical features:
These capture circuit complexity and logic structure. The feature matrix is 108 processes × 5 features, standardized to zero mean and unit variance.
We build pairwise Euclidean distances between processes in the 5-dimensional feature space, then run a Vietoris–Rips filtration and compute persistent homology with Ripser (maxdim=2), with cocycle extraction enabled. Outputs are persistence diagrams for H₀ (connected components), H₁ (loops), and H₂ (voids). Ripser's cocycle extraction returns, for each H₁ feature, a representative cocycle: a set of (process A, process B) pairs whose edges form the cycle in the Vietoris–Rips complex. These pairs identify which processes participate in each topological loop and are used for both biological interpretation (Section 3.2) and visualization (PCA + cocycle edges, Figure 3).
H₀, H₁, H₂: an intuitive ladder. H₀ counts connected components—the number of disconnected pieces. In GLMP, H₀ begins at 108 (one per process) and collapses as the Vietoris–Rips radius grows. H₁ counts loops—closed cycles with no filled-in face bounding them. In a gene regulatory network, this corresponds to a feedback loop; the 33 H₁ features are precisely these unfilled cycles. H₂ counts enclosed voids—analogous to the interior of a hollow sphere. In the cancer GRN literature (Masoomy et al., 2021), H₂ in healthy cells was interpreted as redundant regulatory structures. GLMP yields H₂ = 1. The intuitive ladder: H₀ asks "are the pieces connected?"; H₁ asks "are there feedback loops?"; H₂ asks "are there enclosed cavities?" For GLMP, H₁ is biologically richest.
Mathematical note: Betti numbers and Euler characteristic. The homology groups H₀, H₁, H₂ have ranks β₀, β₁, β₂—the Betti numbers—which count connected components, loops (1-dimensional holes), and voids (2-dimensional holes) respectively. Named for Enrico Betti (1823–1892) and formalized by Henri Poincaré in the 1890s, they provide a geometric view of shape: β₀ counts pieces, β₁ counts independent loops that do not bound any filled region, and β₂ counts enclosed voids. Euler’s formula for connected planar graphs is χ = V − E + F = 2, where F includes the outer (unbounded) face. For example: a triangle has V=3, E=3, F=2 (interior + outer) so χ=2; a tetrahedron has V=4, E=6, F=4 so χ=2; a cube has V=8, E=12, F=6 so χ=2. This generalizes to χ = β₀ − β₁ + β₂ − … via the Betti numbers. In simplicial complexes, faces correspond to 2-simplices—filled triangles. When a closed loop of edges is not the boundary of any 2-simplex (no triangle fills it in), that loop persists as an H₁ feature. Our 33 H₁ loops are precisely those cycles that are not bounded by faces; they are the β₁ contribution to this alternating sum.
Principal component analysis (PCA) finds orthogonal directions of maximum variance in the data. Given a matrix of 108 processes × 5 features, PCA identifies the two directions along which the data vary most; we then project each process onto those two axes. The result is a 2D scatter plot where PC1 (first principal component) captures the largest variance, and PC2 (second) captures the next largest, uncorrelated with PC1. We use PCA for visualization because the Vietoris–Rips complex and H₁ cocycles are computed in full 5D; we need a 2D projection to draw cocycle edges on a plane. PCA preserves as much variance as possible in the projection, so the spatial arrangement in the plot approximates the true distances used for TDA—processes that are close in 5D tend to remain close in the PCA plot, and cycles in 5D appear as cycles in 2D. Alternatives such as t-SNE or UMAP would distort distances and are less suitable when the goal is to faithfully depict topological structure.
We obtain: - H₀: 108 components (one per
process).
- H₁: 33 loops.
- H₂: 1 void.
Why only one H₂? H₂ counts 2-dimensional cavities (e.g., the interior of a hollow tetrahedron). With 108 points in 5D, the Vietoris–Rips complex rarely forms such structures; when it does, the void fills quickly as the radius increases. One short H₂ bar is expected for this data—few points and moderate dimensionality do not typically yield many persistent 2D cavities.
The question we then ask is whether the H₁ loops align with known biology—feedback circuits, stress responses, and regulatory motifs.
To make homology visible, we use two complementary views. First, we project the 5D feature space to 2D using PCA (Section 2.4) and draw cocycle edges—the (process A, process B) pairs that form each H₁ cycle—as colored polygons. We show all five top loops: red = Loop #1, blue = Loop #2, green = Loop #3, purple = Loop #4, orange = Loop #5 (Figure 3). Lac operon, two-component signaling, and SOS are labeled. Second, we build a Mapper graph: cluster nearby processes (n_cubes=12, perc_overlap=0.65), connect overlapping clusters; nodes = clusters (18 nodes), edges = overlaps (45 edges). Cycles in the Mapper graph correspond to H₁ loops (Figure 4). Both visualizations and interactive versions (hover for process names; click nodes to search) are available at the links in Section 5.

Figure 2. Persistence diagram. H₀: 108 components (one per process). H₁: 33 loops. H₂: 1 void.

Figure 3. H₁ loops visualized in 2D (PCA projection). Cocycle edges connect processes that form each cycle. Red = Loop #1, blue = Loop #2, green = Loop #3, purple = Loop #4, orange = Loop #5. PCA projects the 5D feature space to 2D while preserving maximal variance, so distances in the plot approximate true TDA distances.
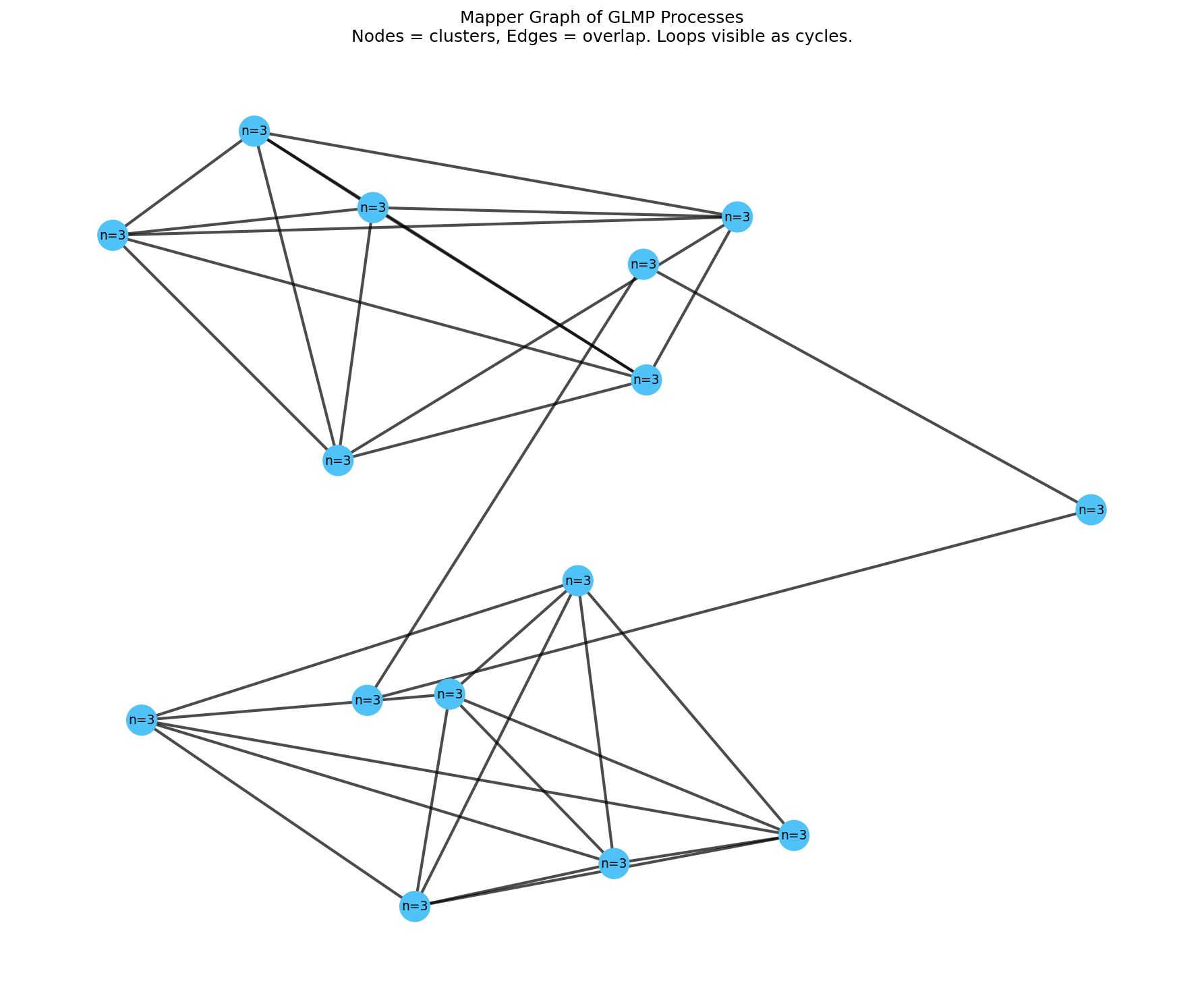
Figure 4. Mapper graph. Nodes = clusters of similar processes; edges = overlapping clusters. Cycles correspond to H₁ loops.
We rank H₁ loops by persistence (death − birth). Highlights:
Loop #1 (persistence 0.563)
10 processes: base excision repair (BER), biofilm formation, BAM complex
assembly, quorum sensing, ribosome assembly, RNA pol recycling, SOS
response, Type III secretion, ubiquitin-proteasome, unfolded protein
response (UPR). Stress response, protein quality control, DNA repair—E.
coli and yeast. Shared “stress + quality control + feedback”
character.
Loop #2 (persistence 0.443)
6 processes: antibiotic efflux pumps, arginine biosynthesis, osmotic
stress response, tryptophan biosynthesis, peroxisome biogenesis,
vacuolar protein sorting. Metabolic regulation and organelle
biogenesis—E. coli and yeast.
Loop #3 (persistence 0.306)
6 processes: biofilm formation, DNA replication elongation, flagellar
assembly, osmotic stress, sigma factor competition, peroxisome
biogenesis. Gene regulation, replication, motility, stress—E. coli and
yeast.
Loop #4 (persistence 0.279)
6 processes: phosphate regulation, translation elongation, translation
termination, tryptophan biosynthesis, osmotic stress response,
sporulation initiation. E. coli, yeast, Bacillus.
Loop #5 (persistence 0.198)
5 processes: ara operon, maltose regulon, Pho regulon, nitrogen
catabolite repression (NCR/TORC1), competence development. Nutrient and
developmental regulation—ara and Pho are classic feedback circuits. E.
coli, yeast, Bacillus.
With the new loop-based feature set, known feedback circuits cluster coherently: SOS, quorum sensing, biofilm in Loop #1 (stress + feedback); ara and Pho in Loop #5 (nutrient-sensing feedback); trp biosynthesis in Loops #2 and #4. Topology recovers regulatory structure—stress, protein quality, nutrient regulation—from structural features alone.
All top five loops mix organisms. Loop #1, #2, #3: E. coli and yeast. Loop #4 and #5: E. coli, yeast, and Bacillus. Topology groups by circuit structure, not by species—regulatory logic transcends organism boundaries.
We reran TDA dropping one feature at a time. Baseline (all five features) gave coherence 0.75 (6 of 8 reference circuits in the top five H₁ loops) and 33 H₁ loops. Ablation results:
| Condition | Coherence | Delta | H₁ loops |
|---|---|---|---|
| Baseline (all 5) | 0.750 | — | 33 |
| Drop node_count | 0.125 | −0.625 | 33 |
| Drop conditional_count | 0.250 | −0.500 | 32 |
| Drop or_gates | 0.375 | −0.375 | 34 |
| Drop and_gates | 0.500 | −0.250 | 32 |
| Drop loops | 0.250 | −0.500 | 27 |
Removal of node_count produced the largest coherence decrease (delta = −0.625), identifying it as the most biologically informative feature. Dropping conditional_count or loops also strongly reduced coherence (−0.500). No single feature is dispensable; the signal is distributed, with node count and conditional/loop structure carrying the most weight.
We randomly permuted circuit labels and recomputed coherence over 1,000 permutations. Observed coherence 0.750 lay well above the null distribution (null mean ± SD: 0.339 ± 0.167; 95th percentile: 0.625; 99th percentile: 0.750). The one-tailed p-value was 0.022 (n = 1,000). Biological coherence at this level is therefore unlikely to arise by chance; the result is statistically significant at p < 0.05.
The appearance of SOS response, ara operon, Pho regulon, and trp biosynthesis in coherent H₁ loops supports the view that TDA on structural features reflects regulatory logic. Loop #1 aggregates stress and protein-quality circuits; Loop #5 groups nutrient-sensing feedback. Using loop (back-edge) features instead of NOT gates yields richer persistence values. The same chart that was infeasible to produce at scale in 1995 can now be generated in seconds; applying TDA to many such flowcharts reveals that feedback loops appear as loops in homology.
Sample size: 108 processes is enough to reveal structure but scaling to 200–500+ is a priority for robustness.
Feature sensitivity: We use five structural features (nodes, conditionals, OR gates, AND gates, loops). Ablation (Section 3.5) shows that node count is the most load-bearing feature; conditional_count and loops also contribute strongly. The result is not an artifact of a single feature. Graph-theoretic enrichment (cycle rank, longest path, gate ratios) is planned for the next pipeline iteration.
Flowcharts: Diagrams are LLM-generated and require fact-checking. The GLMP viewer feedback mechanism supports community validation.
Open question: Does topology predict regulatory function or correlate with known biology? The coherence check supports the latter; prediction would require prospective validation with domain experts.
Validation and robustness: Feature ablation (Section 3.5) and null-model permutation (Section 3.6) are complete. Ablation shows that coherence is distributed across features, with node count most informative. The null model gives p = 0.022 (n = 1,000 permutations)—observed coherence rarely arises by chance. Both results support that the topology is capturing biologically meaningful structure rather than feature-space artifact.
Mapper: We have implemented Mapper on the GLMP feature space (n_cubes=12, perc_overlap=0.65; 18 nodes, 45 edges). An interactive version allows users to click nodes to see constituent processes and search by name. Next: treat circuit classes as nodes for distinct regulatory families; explore persistent cohomology for circular coordinates that might align with “feedback depth” or cascade structure.
Scaling: Expand to 200–500+ genetic circuits.
Topology and sequence (second act). A natural next step is to ask whether topological neighborhoods in GLMP feature space predict shared regulatory sequence motifs—the physical implementation of AND and OR logic on the chromosome. AND/OR in flowcharts correspond to binding-site logic (e.g. dual binding for AND; alternative sites for OR). If circuits in the same H₁ loop share sequence motifs enriched in their promoter regions (e.g. via RegulonDB, YEASTRACT, and motif discovery), topology would be doing something predictive about biology rather than merely descriptive. Cross-organism loops (Loop #1, Loop #5) are the strongest test case, since organism-level confounding cannot explain shared motifs. This would extend the current methodological foundation toward a falsifiable hypothesis: topological neighborhoods are predictive of shared regulatory sequence motifs that implement the logical operations represented in the flowcharts.
Collaboration: We seek biologist validation of flowcharts and interpretations. Jordan Matuszewski and the CUNY Graduate Center TDA seminar group have provided feedback.
We applied TDA to 108 genetic regulatory circuits encoded as Mermaid Markdown flowcharts. The flowcharts are produced with LLM assistance from textual descriptions—the same Lac/β-galactosidase idea that was first sketched in 1995 can now be generated at scale in seconds. With the loop-based feature set, the most persistent H₁ loops correspond to stress and protein-quality circuits (Loop #1: SOS, quorum sensing, biofilm, UPR), metabolic and organelle biogenesis (Loops #2, #3), and nutrient-sensing feedback (Loop #5: ara, Pho, maltose). Topology groups processes by regulatory logic across organisms. The work demonstrates a pipeline: text → visual data → features → topology, and suggests that TDA on structural features captures genuine regulatory architecture. Code, data, and documentation are open source.
We thank Jordan Matuszewski and the CUNY Graduate Center TDA seminar group for feedback. We acknowledge the foundational work of Kevin Gardner and colleagues (ASRC, CCNY) on two-component signaling and kinase/effector regulatory logic.
Carlsson, G., & Vejdemo-Johansson, M. (2021). Topological Data Analysis with Applications. Cambridge University Press. https://doi.org/10.1017/9781108975704
Bauer, U. (2021). Ripser: efficient computation of Vietoris–Rips persistence barcodes. Journal of Applied and Computational Topology 5, 391–423. https://doi.org/10.1007/s41468-021-00071-5
Berg, P., & Singer, M. (1992). Dealing With Genes: The Language of Heredity. University Science Books.
Masoomy, H., Askari, B., Tajik, S., Rizi, A. K., & Jafari, G. R. (2021). Topological analysis of interaction patterns in cancer-specific gene regulatory network: persistent homology approach. Scientific Reports 11, 16414. https://doi.org/10.1038/s41598-021-94847-5
Rivera-Cancel, G., Ko, W. H., Tomchick, D. R., Correa, F., & Gardner, K. H. (2014). Full-length structure of a monomeric histidine kinase reveals basis for sensory regulation. Proceedings of the National Academy of Sciences 111(50), 17839–17844. https://doi.org/10.1073/pnas.1413983111
Swingle, D., Epstein, L., Aymon, R., Isiorho, E. A., Abzalimov, R. R., Favaro, D. C., & Gardner, K. H. (2025). Variations in kinase and effector signaling logic in a bacterial two component signaling network. Journal of Biological Chemistry 301, 108534. https://doi.org/10.1016/j.jbc.2025.108534
Tralie, C., Saul, N., & Bar-On, R. (2018). Ripser.py: A lean persistent homology library for Python. Journal of Open Source Software 3(29), 925. https://doi.org/10.21105/joss.00925
Welz, G. (1995). Is the genome like a computer program? The X Advisor (July 1995). Archived at https://web.archive.org/web/19970310064130/http://landru.unx.com/DD/advisor/docs/jul95/welz.genome0.shtml