Data Cleaning Small Intestine
Ann Wells
April 23, 2023
tdata.FPKM.sample.info <- readRDS(here("Data","20190406_RNAseq_B6_4wk_2DG_counts_phenotypes.RData"))
tdata.FPKM <- readRDS(here("Data","20190406_RNAseq_B6_4wk_2DG_counts_numeric.RData"))
log.tdata.FPKM <- log(tdata.FPKM + 1)
log.tdata.FPKM <- as.data.frame(log.tdata.FPKM)
log.tdata.FPKM.sample.info <- cbind(log.tdata.FPKM, tdata.FPKM.sample.info[,27238:27240])
log.tdata.FPKM.sample.info <- log.tdata.FPKM.sample.info %>% rownames_to_column() %>% filter(rowname != "A113") %>% column_to_rownames()
log.tdata.FPKM.subset <- log.tdata.FPKM[,colMeans(log.tdata.FPKM != 0) > 0.5]
log.tdata.FPKM.sample.info.subset <- cbind(log.tdata.FPKM.subset,tdata.FPKM.sample.info[,27238:27240])
log.tdata.FPKM.sample.info.subset <- log.tdata.FPKM.sample.info.subset %>% rownames_to_column() %>% filter(rowname != "A113") %>% column_to_rownames()
log.tdata.FPKM.sample.info.subset.small.intestine <- log.tdata.FPKM.sample.info.subset %>% rownames_to_column() %>% filter(Tissue == "Small Intestine") %>% column_to_rownames()Wrangle Data
I will use Mouse IDs, Treatment, and Time to keep track of the values in the matrices. All other covariates will be discarded.
# Set rownames by mouse ID and tissue
rownames(log.tdata.FPKM.sample.info.subset.small.intestine) <- paste0(rownames(log.tdata.FPKM.sample.info.subset.small.intestine),":", log.tdata.FPKM.sample.info.subset.small.intestine$Time, ":", log.tdata.FPKM.sample.info.subset.small.intestine$Treatment)
# Discard covariates from columns 17333-17336
log.tdata.FPKM.sample.info.subset.small.intestine <- log.tdata.FPKM.sample.info.subset.small.intestine[,-(17333:17335)]
head(log.tdata.FPKM.sample.info.subset.small.intestine[,1:5])## ENSMUSG00000000001 ENSMUSG00000000028 ENSMUSG00000000031
## A006:4 wks:None 3.984158 1.754404 0.16551444
## A015:96 hrs:2DG 4.654532 1.680828 0.06765865
## A024:4 wks:2DG 4.597037 1.930071 0.12221763
## A033:4 wks:2DG 4.557659 1.821318 0.27763174
## A042:4 wks:None 4.325721 1.912501 0.11332869
## A051:4 wks:None 4.350020 1.888584 0.36464311
## ENSMUSG00000000037 ENSMUSG00000000049
## A006:4 wks:None 0.1484200 0.7839015
## A015:96 hrs:2DG 0.3646431 0.5306283
## A024:4 wks:2DG 1.0152307 1.3480731
## A033:4 wks:2DG 0.1484200 0.9707789
## A042:4 wks:None 0.1043600 1.1052568
## A051:4 wks:None 0.2926696 0.9122827Check Data for Missing Values
WGCNA will have poor results if the data have too many missing values. I checked if any metabolites fall into this category.
log.tdata.FPKM.sample.info.subset.small.intestine.missing <- missing(log.tdata.FPKM.sample.info.subset.small.intestine)
cat("logFPKM: ", goodSamplesGenes(log.tdata.FPKM.sample.info.subset.small.intestine.missing, verbose=0)$allOK, "\n")## logFPKM: TRUEWGCNA reports that all data are good! I now use hierarchical clustering to detect any obvious outliers. I did not see any particularly egregious outliers.
sampleclustering(log.tdata.FPKM.sample.info.subset.small.intestine.missing)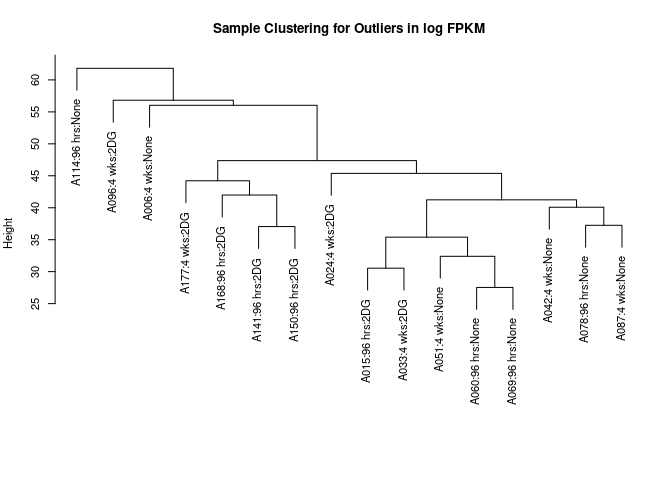
saveRDS(log.tdata.FPKM.sample.info.subset.small.intestine.missing, here("Data","Small Intestine","log.tdata.FPKM.sample.info.subset.small.intestine.missing.WGCNA.RData"))Analysis performed by Ann Wells
The Carter Lab The Jackson Laboratory 2023
ann.wells@jax.org