Pathway Analysis Skeletal Muscle, Small Intestine, and Spleen
Ann Wells
April 23, 2023
Introduction
This dataset contains nine tissues (heart, hippocampus, hypothalamus, kidney, liver, prefrontal cortex, skeletal muscle, small intestine, and spleen) from C57BL/6J mice that were fed 2-deoxyglucose (6g/L) through their drinking water for 96hrs or 4wks. 96hr mice were given their 2DG treatment 2 weeks after the other cohort started the 4 week treatment. The organs from the mice were harvested and processed for metabolomics and transcriptomics. The data in this document pertains to the transcriptomics data only. The counts that were used were FPKM normalized before being log transformed. It was determined that sample A113 had low RNAseq quality and through further analyses with PCA, MA plots, and clustering was an outlier and will be removed for the rest of the analyses performed. This document will determine which pathways are significantly altered by each module.
needed.packages <- c("tidyverse", "here", "functional", "gplots", "dplyr", "GeneOverlap", "R.utils", "reshape2","magrittr","data.table", "RColorBrewer","preprocessCore", "ARTool","emmeans", "phia", "gprofiler2", "rlist","plotly","downloadthis")
for(i in 1:length(needed.packages)){library(needed.packages[i], character.only = TRUE)}
source(here("source_files","WGCNA_source.R"))Module Bar plot
This bar plot shows the number of genes in each module.
modules<-read.csv(here("Data","SM.SI.spleen","log.tdata.FPKM.sample.info.subset.SM.SI.spleen.WGCNA.module.membership.csv"), header=T)
module_barplot(modules)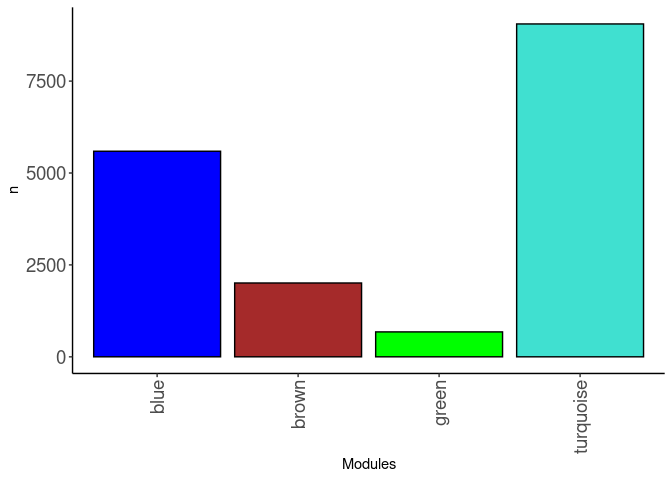
Pathway Analysis
Pathway analysis was performed using the gprofiler package. Genes associated with each module were compared against KEGG and REACTOME databases. Modules that did not contain any significant pathways are blank.
Pathway plots
WGCNA<-read.table(here("Data","SM.SI.spleen","log.tdata.FPKM.sample.info.subset.SM.SI.spleen.WGCNA.module.membership.csv"), header=T)
Data_setup(WGCNA, file = "Annotated_genes_in_SM_SI_spleen_WGCNA_Chang_B6_96hr_4wk.RData", folder="SM.SI.spleen")
Matched<-readRDS(here("Data","SM.SI.spleen","Annotated_genes_in_SM_SI_spleen_WGCNA_Chang_B6_96hr_4wk.RData"))
pathways(Matched, pathwayfile = "Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_SM_SI_spleen_WGCNA.RData", genefile = "Chang_B6_96hr_4wk_gprofiler_gene_annotation_list_SM_SI_spleen_WGCNA.RData", folder = "SM.SI.spleen")
WGCNA.pathway <-readRDS(here("Data","SM.SI.spleen","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_SM_SI_spleen_WGCNA.RData"))blue
brown
green
turquoise
Table of Pathways
WGCNA.pathway <- readRDS(here("Data","SM.SI.spleen","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_SM_SI_spleen_WGCNA.RData"))Blue
if(class(WGCNA.pathway[[1]]) == "numeric"){
print("No pathways significantly overrepresented")
} else {DT.table.path(WGCNA.pathway[[1]][c(11,3:6)])}Brown
if(class(WGCNA.pathway[[2]]) == "numeric"){
print("No pathways significantly overrepresented")
} else {DT.table.path(WGCNA.pathway[[2]][c(11,3:6)])}Green
if(class(WGCNA.pathway[[3]]) == "numeric"){
print("No pathways significantly overrepresented")
} else {DT.table.path(WGCNA.pathway[[3]][c(11,3:6)])}Turquoise
if(class(WGCNA.pathway[[4]]) == "numeric"){
print("No pathways significantly overrepresented")
} else {DT.table.path(WGCNA.pathway[[4]][c(11,3:6)])}Pathway Frequency
WGCNA.pathway <- readRDS(here("Data","SM.SI.spleen","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_SM_SI_spleen_WGCNA.RData"))
#pdf("Counts_of_each_pathway_identified_within_SM.SI.spleen.pdf")
count <- count.pathways(WGCNA.pathway)[1] 613 [1] 573
#dev.off()Frequency Table
DT.table.freq(count)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=count,aes(x=list.pathways,y=Freq))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(count[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=count$list.pathways) + xlab("Modules")
print(p)
#dev.off()Analysis performed by Ann Wells
The Carter Lab The Jackson Laboratory 2023
ann.wells@jax.org