Comparing Significant Overlaps (Jaccard) of Genes and Pathways for Modules Across Tissues of Interest
Ann Wells
April 23, 2023
Introduction and Data files
This dataset contains nine tissues (heart, hippocampus, hypothalamus, kidney, liver, prefrontal cortex, skeletal muscle, small intestine, and spleen) from C57BL/6J mice that were fed 2-deoxyglucose (6g/L) through their drinking water for 96hrs or 4wks. 96hr mice were given their 2DG treatment 2 weeks after the other cohort started the 4 week treatment. The organs from the mice were harvested and processed for metabolomics and transcriptomics. The data in this document pertains to the transcriptomics data only. The counts that were used were FPKM normalized before being log transformed. It was determined that sample A113 had low RNAseq quality and through further analyses with PCA, MA plots, and clustering was an outlier and will be removed for the rest of the analyses performed. This document will look at modules that were significantly correlated with traits and had pathways identified within the WGCNA. Those modules will be assessed to see which modules from other tissues and their modules significantly overlapped.
needed.packages <- c("tidyverse", "here", "functional", "gplots", "dplyr", "GeneOverlap", "R.utils", "reshape2","magrittr","data.table", "RColorBrewer","preprocessCore", "ARTool","emmeans", "phia", "gprofiler2", "rlist", "pheatmap")
for(i in 1:length(needed.packages)){library(needed.packages[i], character.only = TRUE)}
source(here("source_files","WGCNA_source.R"))heart <- readRDS(here("Data","Heart","log.tdata.FPKM.sample.info.subset.heart.WGCNA.module.membership.RData"))
hip <- readRDS(here("Data","Hippocampus","log.tdata.FPKM.sample.info.subset.hippocampus.WGCNA.module.membership.RData"))
hyp <- readRDS(here("Data","Hypothalamus","log.tdata.FPKM.sample.info.subset.hypothalamus.WGCNA.module.membership.RData"))
kidney <- readRDS(here("Data","Kidney","log.tdata.FPKM.sample.info.subset.kidney.WGCNA.module.membership.RData"))
liver <- readRDS(here("Data","Liver","log.tdata.FPKM.sample.info.subset.liver.WGCNA.module.membership.RData"))
cortex <- readRDS(here("Data","Prefrontal Cortex","log.tdata.FPKM.sample.info.subset.prefrontal.cortex.WGCNA.module.membership.RData"))
muscle <- readRDS(here("Data","Skeletal Muscle","log.tdata.FPKM.sample.info.subset.skeletal.muscle.WGCNA.module.membership.RData"))
small.intestine <- readRDS(here("Data","Small Intestine","log.tdata.FPKM.sample.info.subset.small.intestine.WGCNA.module.membership.RData"))
spleen <- readRDS(here("Data","Spleen","log.tdata.FPKM.sample.info.subset.spleen.WGCNA.module.membership.RData"))Jaccard Plots
Genes
The modules that were chosen according to the filtering criteria described in the paper were compared within the jaccard index across all other tissues. The significant overlaps indicate which modules have a significant number of shared genes.
## heart
Matched.module.heart <- split(heart$Gene, heart$Module)
modulenames <- c()
for(i in 1:length(Matched.module.heart)){
modules <- unique(heart$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.heart) <- modulenames
## hippocampus
Matched.module.hip <- split(hip$Gene, hip$Module)
modulenames <- c()
for(i in 1:length(Matched.module.hip)){
modules <- unique(hip$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.hip) <- modulenames
## hypothalamus
Matched.module.hyp <- split(hyp$Gene, hyp$Module)
modulenames <- c()
for(i in 1:length(Matched.module.hyp)){
modules <- unique(hyp$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.hyp) <- modulenames
## kidney
Matched.module.kidney <- split(kidney$Gene, kidney$Module)
modulenames <- c()
for(i in 1:length(Matched.module.kidney)){
modules <- unique(kidney$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.kidney) <- modulenames
## liver
Matched.module.liver <- split(liver$Gene, liver$Module)
modulenames <- c()
for(i in 1:length(Matched.module.liver)){
modules <- unique(liver$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.liver) <- modulenames
## prefrontal cortex
Matched.module.cortex <- split(cortex$Gene, cortex$Module)
modulenames <- c()
for(i in 1:length(Matched.module.cortex)){
modules <- unique(cortex$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.cortex) <- modulenames
## skeletal muscle
Matched.module.muscle <- split(muscle$Gene, muscle$Module)
modulenames <- c()
for(i in 1:length(Matched.module.muscle)){
modules <- unique(muscle$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.muscle) <- modulenames
## small intestine
Matched.module.small.intestine <- split(small.intestine$Gene, small.intestine$Module)
modulenames <- c()
for(i in 1:length(Matched.module.small.intestine)){
modules <- unique(small.intestine$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.small.intestine) <- modulenames
## spleen
Matched.module.spleen <- split(spleen$Gene, spleen$Module)
modulenames <- c()
for(i in 1:length(Matched.module.spleen)){
modules <- unique(spleen$Module)
name <- str_split(modules,"_")
modulenames[i] <- name[[i]][2]
#gene <- as.vector(Matched.module[[i]])
}
modulenames <- sort(modulenames)
names(Matched.module.spleen) <- modulenamestissues.treat <- list("Heart" = Matched.module.heart$darkgreen, "Liver" = Matched.module.liver$darkolivegreen, "Hypothalamus" = Matched.module.hyp$bisque4, "Prefrontal Cortex" = Matched.module.cortex$bisque2, "Muscle" = Matched.module.muscle$salmon2, "Small Intestine" = Matched.module.small.intestine$green4)mods<-crossprod(table(stack(tissues.treat)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(tissues.treat[rownames(mods)[i]]),unlist(tissues.treat[colnames(mods)[j]])))
S <- I/(length(unlist(tissues.treat[rownames(mods)[i]]))+length(unlist(tissues.treat[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(tissues.treat[rownames(mods)[i]])),unique(unlist(tissues.treat[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Fisher's_exact_text_p_val_Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"))
write.csv(Jaccard,here("Data","Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"))pval <- read.csv(here("Data","Fisher's_exact_text_p_val_Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,.07,length.out = 100)
pheatmap(mods,cluster_cols=F,cluster_rows=F, fontsize = 10, fontsize_row = 12,fontsize_col = 12,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)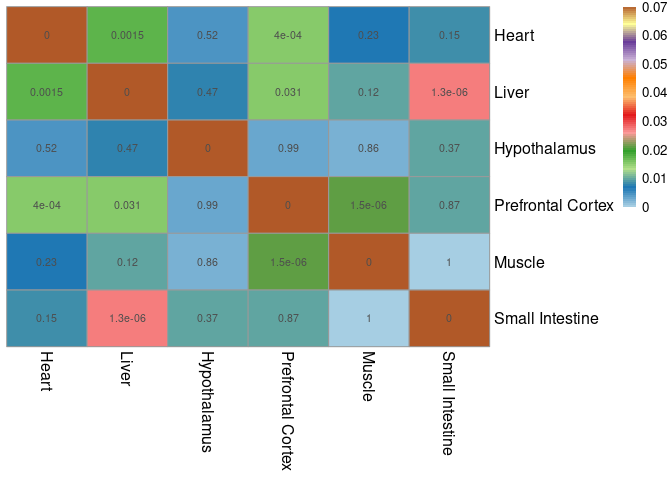
Pathways
The modules that were chosen according to the filtering criteria described in the paper were compared within the jaccard index across all other tissues. The significant overlaps indicate which modules have a significant number of shared pathways.
heart <- readRDS(here("Data","Heart","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_heart_WGCNA.RData"))
hip <- readRDS(here("Data","Hippocampus","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_hippocampus_WGCNA.RData"))
hyp <- readRDS(here("Data","Hypothalamus","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_hypothalamus_WGCNA.RData"))
kidney <- readRDS(here("Data","Kidney","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_kidney_WGCNA.RData"))
liver <- readRDS(here("Data","Liver","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_liver_WGCNA.RData"))
cortex <- readRDS(here("Data","Prefrontal Cortex","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_prefrontal_cortex_WGCNA.RData"))
muscle <- readRDS(here("Data","Skeletal Muscle","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_skeletal_muscle_WGCNA.RData"))
small.intestine <- readRDS(here("Data","Small Intestine","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_small_intestine_WGCNA.RData"))
spleen <- readRDS(here("Data","Spleen","Chang_B6_96hr_4wk_gprofiler_pathway_annotation_list_spleen_WGCNA.RData"))tissues.treat <- list("Heart" = Matched.module.heart$darkgreen, "Liver" = Matched.module.liver$darkolivegreen, "Hypothalamus" = Matched.module.hyp$bisque4, "Prefrontal Cortex" = Matched.module.cortex$bisque2, "Muscle" = Matched.module.muscle$salmon2, "Small Intestine" = Matched.module.small.intestine$green4)mods<-crossprod(table(stack(tissues.treat)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(tissues.treat[rownames(mods)[i]]),unlist(tissues.treat[colnames(mods)[j]])))
S <- I/(length(unlist(tissues.treat[rownames(mods)[i]]))+length(unlist(tissues.treat[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(tissues.treat[rownames(mods)[i]])),unique(unlist(tissues.treat[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"))
write.csv(Jaccard,here("Data","Jaccard_Index_module_C57BL6J_treat_across_tissues_pathways.csv"))Jaccard <- read.csv(here("Data","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"), row.names = 1)
pval <- read.csv(here("Data","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_treat_across_tissues.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,1,length.out = 100)
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 10, fontsize_row = 12,fontsize_col = 12,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)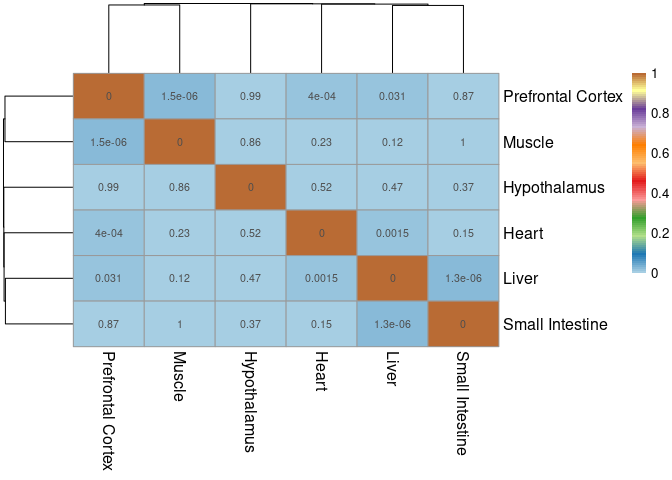
Jaccard Plots of Pathways within Each Tissue
Pathways for each module that was chosen according to the filtering criteria described in the paper were compared to determine if there was significant overlap of genes across pathways identified within each tissue. Liver is missing because only one pathway was identified.
Heart
heart.darkgreen <- heart[[4]][,c(11,16)]
heart.darkgreen <- as.list(heart.darkgreen)
heart.intersection <- as.list(heart.darkgreen$intersection)
names(heart.intersection) <- heart.darkgreen$term_name
heart.darkgreen <- heart.intersection
small.intestine.green4 <- small.intestine[[18]]
small.intestine.green4 <- as.list(small.intestine.green4)
small.intestine.intersection <- as.list(small.intestine.green4$intersection)
names(small.intestine.intersection) <- small.intestine.green4$term_name
small.intestine.green4 <- small.intestine.intersection
cortex.bisque2 <- cortex[[3]][,c(11,16)]
cortex.bisque2 <- as.list(cortex.bisque2)
cortex.intersection <- as.list(cortex.bisque2$intersection)
names(cortex.intersection) <- cortex.bisque2$term_name
cortex.bisque2 <- cortex.intersection
hyp.bisque4 <- hyp[[3]][,c(11,16)]
hyp.bisque4 <- as.list(hyp.bisque4)
hyp.intersection <- as.list(hyp.bisque4$intersection)
names(hyp.intersection) <- hyp.bisque4$term_name
hyp.bisque4 <- hyp.intersection
muscle.salmon2 <- muscle[[35]][,c(11,16)]
muscle.salmon2 <- as.list(muscle.salmon2)
muscle.intersection <- as.list(muscle.salmon2$intersection)
names(muscle.intersection) <- muscle.salmon2$term_name
muscle.salmon2 <- muscle.intersection
liver.darkolivegreen <- liver[[8]][,c(11,16)]
liver.darkolivegreen <- as.list(liver.darkolivegreen)
liver.intersection <- as.list(liver.darkolivegreen$intersection)
names(liver.intersection) <- liver.darkolivegreen$term_name
liver.darkolivegreen <- liver.intersectionmods<-crossprod(table(stack(heart.darkgreen)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(heart.darkgreen[rownames(mods)[i]]),unlist(heart.darkgreen[colnames(mods)[j]])))
S <- I/(length(unlist(heart.darkgreen[rownames(mods)[i]]))+length(unlist(heart.darkgreen[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(heart.darkgreen[rownames(mods)[i]])),unique(unlist(heart.darkgreen[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Heart","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_heart.csv"))
write.csv(Jaccard,here("Data","Heart","Jaccard_Index_module_C57BL6J_genes_across_pathways_heart.csv"))pval <- read.csv(here("Data","Heart","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_heart.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,1,length.out = 100)
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 7, fontsize_row = 10,fontsize_col = 7,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)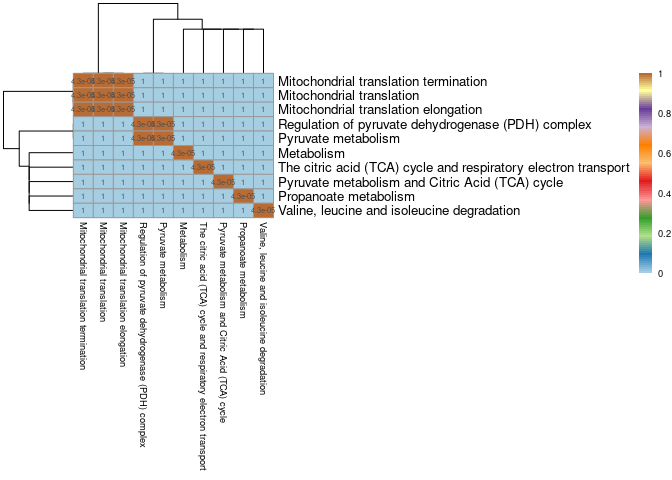
Hypothalamus
mods<-crossprod(table(stack(hyp.bisque4)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(hyp.bisque4[rownames(mods)[i]]),unlist(hyp.bisque4[colnames(mods)[j]])))
S <- I/(length(unlist(hyp.bisque4[rownames(mods)[i]]))+length(unlist(hyp.bisque4[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(hyp.bisque4[rownames(mods)[i]])),unique(unlist(hyp.bisque4[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Hypothalamus","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_hypothalamus.csv"))
write.csv(Jaccard,here("Data","Hypothalamus","Jaccard_Index_module_C57BL6J_genes_across_pathways_hypothalamus.csv"))pval <- read.csv(here("Data","Hypothalamus","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_hypothalamus.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,1,length.out = 100)
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 10, fontsize_row = 6,fontsize_col = 6,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)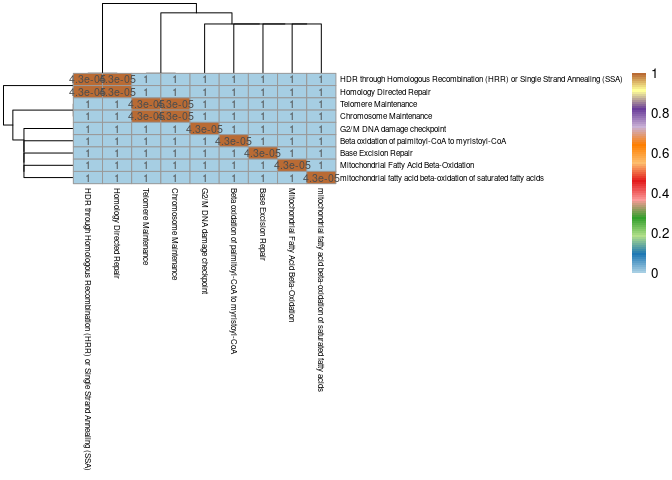
Prefrontal Cortex
mods<-crossprod(table(stack(cortex.bisque2)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(cortex.bisque2[rownames(mods)[i]]),unlist(cortex.bisque2[colnames(mods)[j]])))
S <- I/(length(unlist(cortex.bisque2[rownames(mods)[i]]))+length(unlist(cortex.bisque2[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(cortex.bisque2[rownames(mods)[i]])),unique(unlist(cortex.bisque2[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Prefrontal Cortex","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_cortex.csv"))
write.csv(Jaccard,here("Data","Prefrontal Cortex","Jaccard_Index_module_C57BL6J_genes_across_pathways_cortex.csv"))pval <- read.csv(here("Data","Prefrontal Cortex","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_cortex.csv"), row.names = 1)
pval2 <- signif(pval, 1)
breaks<-seq(0,1,length.out = 100)
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 10, fontsize_row = 10,fontsize_col = 7,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)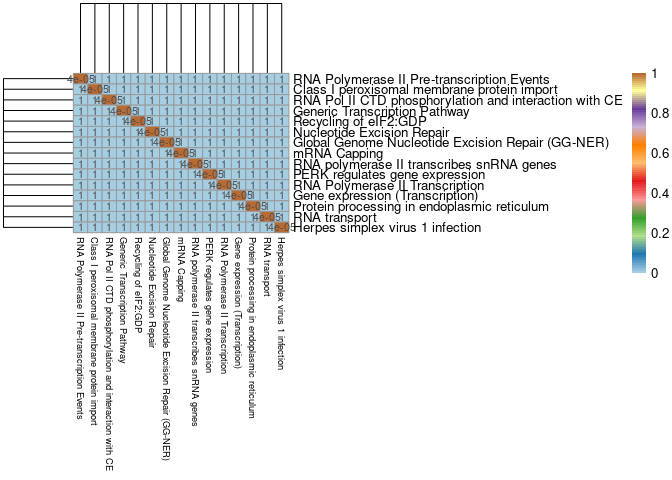
Small Intestine
mods<-crossprod(table(stack(small.intestine.green4)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(small.intestine.green4[rownames(mods)[i]]),unlist(small.intestine.green4[colnames(mods)[j]])))
S <- I/(length(unlist(small.intestine.green4[rownames(mods)[i]]))+length(unlist(small.intestine.green4[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(small.intestine.green4[rownames(mods)[i]])),unique(unlist(small.intestine.green4[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Small Intestine","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_small.intestine.csv"))
write.csv(Jaccard,here("Data","Small Intestine","Jaccard_Index_module_C57BL6J_genes_across_pathways_small.intestine.csv"))pval <- read.csv(here("Data","Small Intestine","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_small.intestine.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,1,length.out = 100)
#pdf("Small Intestine Jaccard Pathways.pdf")
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 7, fontsize_row = 3.5,fontsize_col = 3,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), breaks = breaks)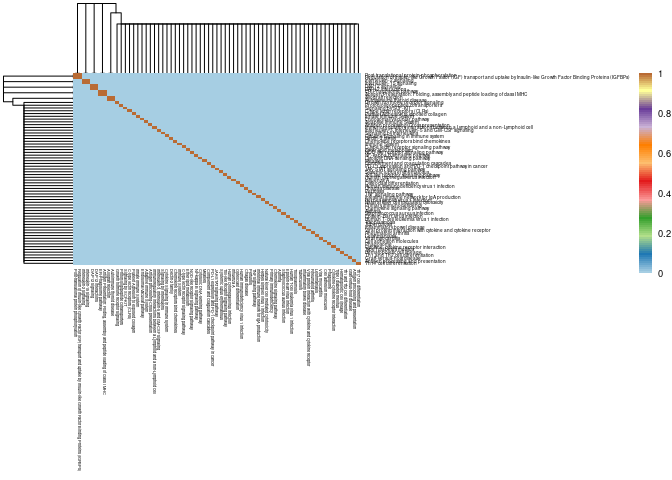
#dev.off()Skeletal Muscle
mods<-crossprod(table(stack(muscle.salmon2)))
for (i in 1:length(rownames(mods))) {
for (j in 1:length(colnames(mods))) {
I <- length(intersect(unlist(muscle.salmon2[rownames(mods)[i]]),unlist(muscle.salmon2[colnames(mods)[j]])))
S <- I/(length(unlist(muscle.salmon2[rownames(mods)[i]]))+length(unlist(muscle.salmon2[colnames(mods)[j]]))-I)
mods[i,j]<-S
}
} matrix<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
p.val<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
Jaccard<-matrix(NA,nrow = nrow(mods),ncol = ncol(mods))
for(i in 1:length(rownames(mods))){
for(j in 1:length(colnames(mods))){
overlap<-newGeneOverlap(unique(unlist(muscle.salmon2[rownames(mods)[i]])),unique(unlist(muscle.salmon2[colnames(mods)[j]])))
overlap2<-testGeneOverlap(overlap)
Jaccard[i,j]<-getJaccard(overlap2)
p.val[i,j]<-getPval(overlap2)
}
}
colnames(p.val)<-colnames(mods)
rownames(p.val)<-rownames(mods)
colnames(Jaccard)<-colnames(mods)
rownames(Jaccard)<-rownames(mods)
write.csv(p.val,here("Data","Skeletal Muscle","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_muscle.csv"))
write.csv(Jaccard,here("Data","Skeletal Muscle","Jaccard_Index_module_C57BL6J_genes_across_pathways_muscle.csv"))pval <- read.csv(here("Data","Skeletal Muscle","Fisher's_exact_text_p_val_pathways_Jaccard_Index_module_C57BL6J_genes_across_pathways_muscle.csv"), row.names = 1)
pval2 <- signif(pval, 2)
breaks<-seq(0,1,length.out = 100)
pheatmap(mods,cluster_cols=T,cluster_rows=T, fontsize = 10, fontsize_row = 7,fontsize_col = 4.5,color = colorRampPalette(brewer.pal(n = 12, name = "Paired"))(100), display_numbers = pval2, breaks = breaks)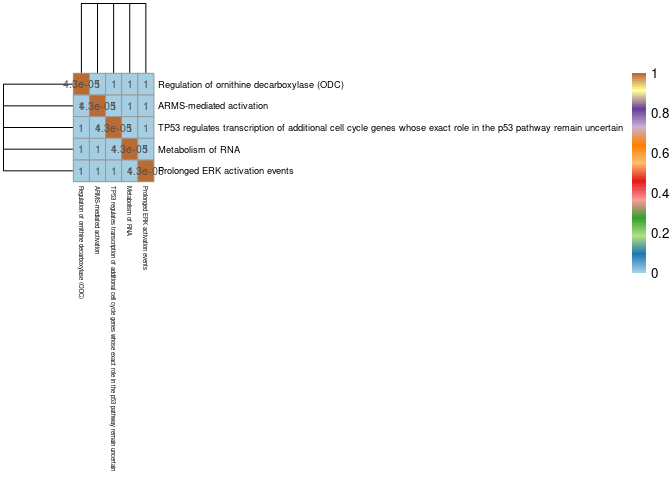
Analysis performed by Ann Wells
The Carter Lab The Jackson Laboratory 2023
ann.wells@jax.org