Comparing Significant Overlaps (Jaccard) for Modules Related to Traits through Pathways Identified
Ann Wells
April 23, 2023
Introduction and Data files
options(stringsAsFactors = F)
knitr::opts_chunk$set(echo = TRUE, warning = FALSE, message = FALSE)
knitr::opts_knit$set(progress=FALSE)This dataset contains nine tissues (heart, hippocampus, hypothalamus, kidney, liver, prefrontal cortex, skeletal muscle, small intestine, and spleen) from C57BL/6J mice that were fed 2-deoxyglucose (6g/L) through their drinking water for 96hrs or 4wks. 96hr mice were given their 2DG treatment 2 weeks after the other cohort started the 4 week treatment. The organs from the mice were harvested and processed for metabolomics and transcriptomics. The data in this document pertains to the transcriptomics data only. The counts that were used were FPKM normalized before being log transformed. It was determined that sample A113 had low RNAseq quality and through further analyses with PCA, MA plots, and clustering was an outlier and will be removed for the rest of the analyses performed. This document will look at modules that were significantly correlated with traits and had pathways identified within the WGCNA. Those modules will be assessed to see which modules from other tissues and their modules significantly overlapped.
Load in Packages and read in files
needed.packages <- c("tidyverse", "here", "functional", "gplots", "dplyr", "GeneOverlap", "R.utils", "reshape2","magrittr","data.table", "RColorBrewer","preprocessCore", "ARTool","emmeans", "phia", "gprofiler2", "rlist")
for(i in 1:length(needed.packages)){library(needed.packages[i], character.only = TRUE)}
source(here("source_files","WGCNA_source.R"))filtered.pathways <- readRDS(here("Data","Chang_2DG_BL6_gprofiler_annotation_filtered_list_modules.RData"))Tissue
Each tissue module was compared to the traits in the experiment. The modules that were significantly correlated for a trait were then compared within the jaccard index across all other tissues. The significant overlaps were then summarized to determine which pathways were most common. Below are the pathway lists and their frequency for each module within a tissue that was also correlated to a trait and their frequency bar plots.
Heart
Darkgreen
jaccard.heart.darkgreen <- filtered.pathways[which(str_detect(names(filtered.pathways),"Heart_darkgreen"))
]
for(i in 1:length(jaccard.heart.darkgreen)){
tissue <- str_split(names(jaccard.heart.darkgreen), pattern = "_")[[i]][3]
heart <- str_split(names(jaccard.heart.darkgreen), pattern = "_")[[i]][1]
jaccard.heart.darkgreen[[i]] <- jaccard.heart.darkgreen[[i]] %>%
mutate(Heart = if(heart == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.heart.darkgreen.merged <- map_df(jaccard.heart.darkgreen, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.heart.darkgreen.merged <- jaccard.heart.darkgreen.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.heart.darkgreen.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.heart.darkgreen.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.heart.darkgreen.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.heart.darkgreen.merged$Var1) + xlab("Modules")
print(p)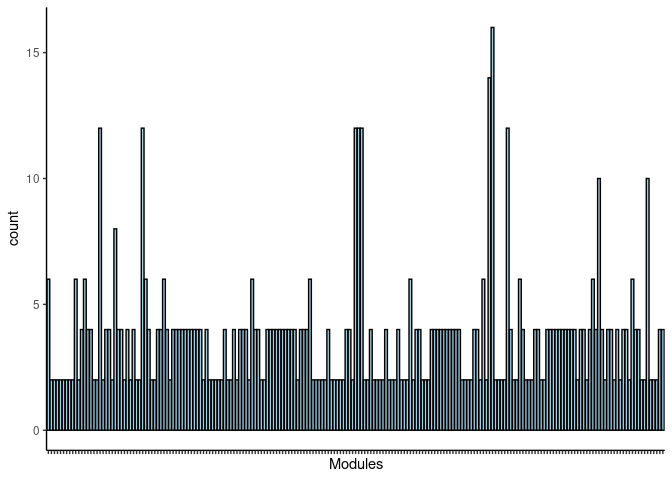
#dev.off()Darkgrey
jaccard.heart.darkgrey <- filtered.pathways[which(str_detect(names(filtered.pathways),"Heart_darkgrey"))
]
for(i in 1:length(jaccard.heart.darkgrey)){
tissue <- str_split(names(jaccard.heart.darkgrey), pattern = "_")[[i]][3]
heart <- str_split(names(jaccard.heart.darkgrey), pattern = "_")[[i]][1]
jaccard.heart.darkgrey[[i]] <- jaccard.heart.darkgrey[[i]] %>%
mutate(Heart = if(heart == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.heart.darkgrey.merged <- map_df(jaccard.heart.darkgrey, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.heart.darkgrey.merged <- jaccard.heart.darkgrey.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.heart.darkgrey.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.heart.darkgrey.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.heart.darkgrey.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.heart.darkgrey.merged$Var1) + xlab("Modules")
print(p)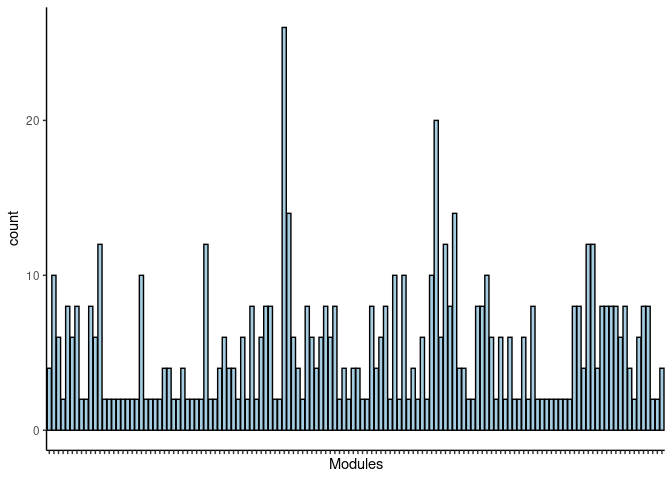
#dev.off()Darkorange
jaccard.heart.darkorange <- filtered.pathways[which(str_detect(names(filtered.pathways),"Heart_darkorange"))
]
for(i in 1:length(jaccard.heart.darkorange)){
tissue <- str_split(names(jaccard.heart.darkorange), pattern = "_")[[i]][3]
heart <- str_split(names(jaccard.heart.darkorange), pattern = "_")[[i]][1]
jaccard.heart.darkorange[[i]] <- jaccard.heart.darkorange[[i]] %>%
mutate(Heart = if(heart == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.heart.darkorange.merged <- map_df(jaccard.heart.darkorange, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.heart.darkorange.merged <- jaccard.heart.darkorange.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.heart.darkorange.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.heart.darkorange.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.heart.darkorange.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.heart.darkorange.merged$Var1) + xlab("Modules")
print(p)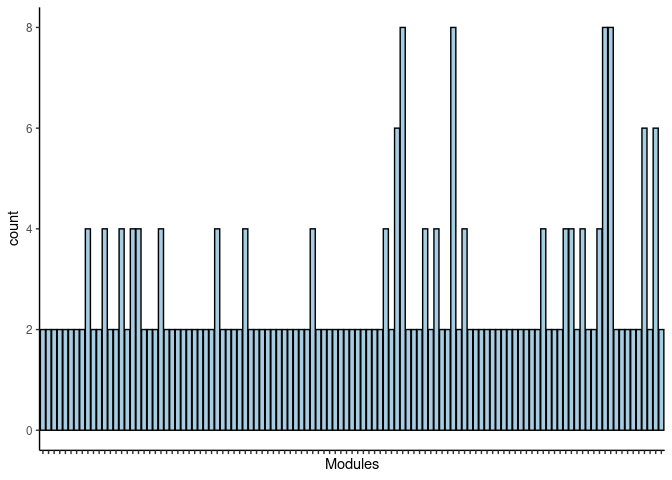
#dev.off()Plum1
jaccard.heart.plum1 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Heart_plum1"))
]
for(i in 1:length(jaccard.heart.plum1)){
tissue <- str_split(names(jaccard.heart.plum1), pattern = "_")[[i]][3]
heart <- str_split(names(jaccard.heart.plum1), pattern = "_")[[i]][1]
jaccard.heart.plum1[[i]] <- jaccard.heart.plum1[[i]] %>%
mutate(Heart = if(heart == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.heart.plum1.merged <- map_df(jaccard.heart.plum1, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.heart.plum1.merged <- jaccard.heart.plum1.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.heart.plum1.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.heart.plum1.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.heart.plum1.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.heart.plum1.merged$Var1) + xlab("Modules")
print(p)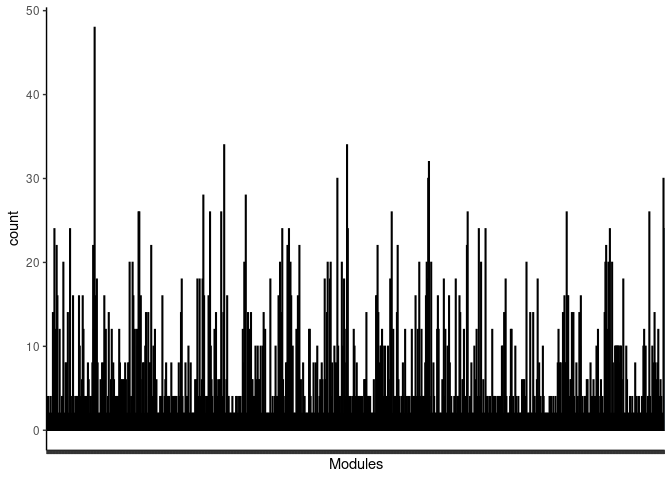
#dev.off()Hippocampus
Green
jaccard.Hippocampus.green <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hippocampus_green"))
]
for(i in 1:length(jaccard.Hippocampus.green)){
tissue <- str_split(names(jaccard.Hippocampus.green), pattern = "_")[[i]][3]
hippocampus <- str_split(names(jaccard.Hippocampus.green), pattern = "_")[[i]][1]
jaccard.Hippocampus.green[[i]] <- jaccard.Hippocampus.green[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(hippocampus == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Hippocampus.green.merged <- map_df(jaccard.Hippocampus.green, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Hippocampus.green.merged <- jaccard.Hippocampus.green.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Hippocampus.green.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Hippocampus.green.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Hippocampus.green.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Hippocampus.green.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Coral1
jaccard.hippocampus.coral1 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hippocampus_coral1"))
]
for(i in 1:length(jaccard.hippocampus.coral1)){
tissue <- str_split(names(jaccard.hippocampus.coral1), pattern = "_")[[i]][3]
hippocampus <- str_split(names(jaccard.hippocampus.coral1), pattern = "_")[[i]][1]
jaccard.hippocampus.coral1[[i]] <- jaccard.hippocampus.coral1[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(hippocampus == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.hippocampus.coral1.merged <- map_df(jaccard.hippocampus.coral1, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.hippocampus.coral1.merged <- jaccard.hippocampus.coral1.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.hippocampus.coral1.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.hippocampus.coral1.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.hippocampus.coral1.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.hippocampus.coral1.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Darkslateblue
jaccard.hippocampus.darkslateblue <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hippocampus_darkslateblue"))
]
for(i in 1:length(jaccard.hippocampus.darkslateblue)){
tissue <- str_split(names(jaccard.hippocampus.darkslateblue), pattern = "_")[[i]][3]
hippocampus <- str_split(names(jaccard.hippocampus.darkslateblue), pattern = "_")[[i]][1]
jaccard.hippocampus.darkslateblue[[i]] <- jaccard.hippocampus.darkslateblue[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(hippocampus == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.hippocampus.darkslateblue.merged <- map_df(jaccard.hippocampus.darkslateblue, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.hippocampus.darkslateblue.merged <- jaccard.hippocampus.darkslateblue.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.hippocampus.darkslateblue.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.hippocampus.darkslateblue.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.hippocampus.darkslateblue.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.hippocampus.darkslateblue.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Darkolivegreen
jaccard.hippocampus.darkolivegreen <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hippocampus_darkolivegreen"))
]
for(i in 1:length(jaccard.hippocampus.darkolivegreen)){
tissue <- str_split(names(jaccard.hippocampus.darkolivegreen), pattern = "_")[[i]][3]
hippocampus <- str_split(names(jaccard.hippocampus.darkolivegreen), pattern = "_")[[i]][1]
jaccard.hippocampus.darkolivegreen[[i]] <- jaccard.hippocampus.darkolivegreen[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(hippocampus == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.hippocampus.darkolivegreen.merged <- map_df(jaccard.hippocampus.darkolivegreen, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.hippocampus.darkolivegreen.merged <- jaccard.hippocampus.darkolivegreen.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.hippocampus.darkolivegreen.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.hippocampus.darkolivegreen.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.hippocampus.darkolivegreen.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.hippocampus.darkolivegreen.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Khaki3
jaccard.hippocampus.khaki3 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hippocampus_khaki3"))
]
for(i in 1:length(jaccard.hippocampus.khaki3)){
tissue <- str_split(names(jaccard.hippocampus.khaki3), pattern = "_")[[i]][3]
hippocampus <- str_split(names(jaccard.hippocampus.khaki3), pattern = "_")[[i]][1]
jaccard.hippocampus.khaki3[[i]] <- jaccard.hippocampus.khaki3[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(hippocampus == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.hippocampus.khaki3.merged <- map_df(jaccard.hippocampus.khaki3, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.hippocampus.khaki3.merged <- jaccard.hippocampus.khaki3.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.hippocampus.khaki3.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.hippocampus.khaki3.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.hippocampus.khaki3.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.hippocampus.khaki3.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Hypothalamus
Midnightblue
jaccard.Hypothalamus.midnightblue <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hypothalamus_midnightblue"))
]
for(i in 1:length(jaccard.Hypothalamus.midnightblue)){
tissue <- str_split(names(jaccard.Hypothalamus.midnightblue), pattern = "_")[[i]][3]
hypothalamus <- str_split(names(jaccard.Hypothalamus.midnightblue), pattern = "_")[[i]][1]
jaccard.Hypothalamus.midnightblue[[i]] <- jaccard.Hypothalamus.midnightblue[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(hypothalamus == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Hypothalamus.midnightblue.merged <- map_df(jaccard.Hypothalamus.midnightblue, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Hypothalamus.midnightblue.merged <- jaccard.Hypothalamus.midnightblue.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Hypothalamus.midnightblue.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Hypothalamus.midnightblue.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Hypothalamus.midnightblue.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Hypothalamus.midnightblue.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Bisque4
jaccard.Hypothalamus.bisque4 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Hypothalamus_bisque4"))
]
for(i in 1:length(jaccard.Hypothalamus.bisque4)){
tissue <- str_split(names(jaccard.Hypothalamus.bisque4), pattern = "_")[[i]][3]
hypothalamus <- str_split(names(jaccard.Hypothalamus.bisque4), pattern = "_")[[i]][1]
jaccard.Hypothalamus.bisque4[[i]] <- jaccard.Hypothalamus.bisque4[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(hypothalamus == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Hypothalamus.bisque4.merged <- map_df(jaccard.Hypothalamus.bisque4, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Hypothalamus.bisque4.merged <- jaccard.Hypothalamus.bisque4.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Hypothalamus.bisque4.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Hypothalamus.bisque4.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Hypothalamus.bisque4.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Hypothalamus.bisque4.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Kidney
Lightyellow
jaccard.Kidney.lightyellow <- filtered.pathways[which(str_detect(names(filtered.pathways),"Kidney_lightyellow"))
]
for(i in 1:length(jaccard.Kidney.lightyellow)){
tissue <- str_split(names(jaccard.Kidney.lightyellow), pattern = "_")[[i]][3]
kidney <- str_split(names(jaccard.Kidney.lightyellow), pattern = "_")[[i]][1]
jaccard.Kidney.lightyellow[[i]] <- jaccard.Kidney.lightyellow[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(kidney == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Kidney.lightyellow.merged <- map_df(jaccard.Kidney.lightyellow, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Kidney.lightyellow.merged <- jaccard.Kidney.lightyellow.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Kidney.lightyellow.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Kidney.lightyellow.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Kidney.lightyellow.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Kidney.lightyellow.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Purple
jaccard.Kidney.purple <- filtered.pathways[which(str_detect(names(filtered.pathways),"Kidney_purple"))
]
for(i in 1:length(jaccard.Kidney.purple)){
tissue <- str_split(names(jaccard.Kidney.purple), pattern = "_")[[i]][3]
kidney <- str_split(names(jaccard.Kidney.purple), pattern = "_")[[i]][1]
jaccard.Kidney.purple[[i]] <- jaccard.Kidney.purple[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(kidney == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Kidney.purple.merged <- map_df(jaccard.Kidney.purple, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Kidney.purple.merged <- jaccard.Kidney.purple.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Kidney.purple.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Kidney.purple.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Kidney.purple.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Kidney.purple.merged$Var1) + xlab("Modules")
print(p)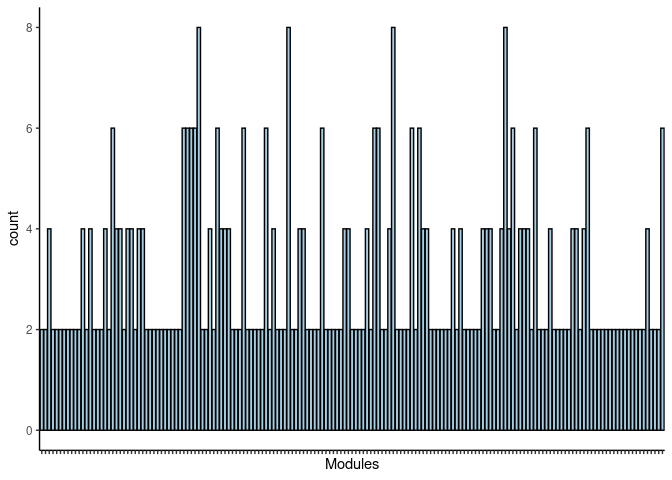
#dev.off()Lightcyan
jaccard.Kidney.lightcyan <- filtered.pathways[which(str_detect(names(filtered.pathways),"Kidney_lightcyan"))
]
for(i in 1:length(jaccard.Kidney.lightcyan)){
tissue <- str_split(names(jaccard.Kidney.lightcyan), pattern = "_")[[i]][3]
kidney <- str_split(names(jaccard.Kidney.lightcyan), pattern = "_")[[i]][1]
jaccard.Kidney.lightcyan[[i]] <- jaccard.Kidney.lightcyan[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(kidney == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Kidney.lightcyan.merged <- map_df(jaccard.Kidney.lightcyan, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Kidney.lightcyan.merged <- jaccard.Kidney.lightcyan.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Kidney.lightcyan.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Kidney.lightcyan.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Kidney.lightcyan.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Kidney.lightcyan.merged$Var1) + xlab("Modules")
print(p)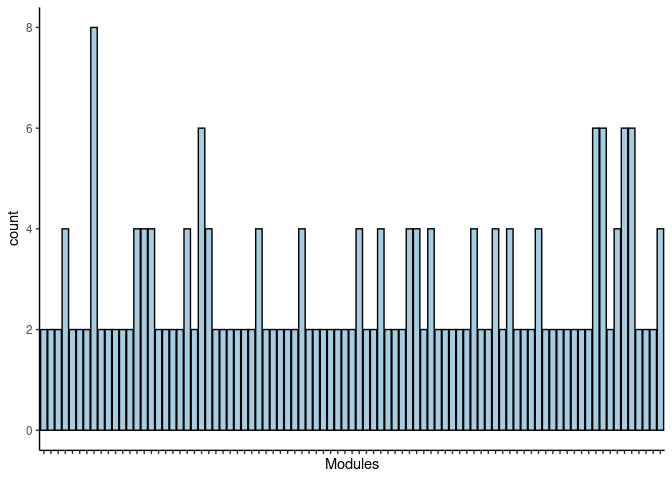
#dev.off()Blue
jaccard.Kidney.blue <- filtered.pathways[which(str_detect(names(filtered.pathways),"Kidney_blue"))
]
for(i in 1:length(jaccard.Kidney.blue)){
tissue <- str_split(names(jaccard.Kidney.blue), pattern = "_")[[i]][3]
kidney <- str_split(names(jaccard.Kidney.blue), pattern = "_")[[i]][1]
jaccard.Kidney.blue[[i]] <- jaccard.Kidney.blue[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(kidney == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Kidney.blue.merged <- map_df(jaccard.Kidney.blue, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Kidney.blue.merged <- jaccard.Kidney.blue.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Kidney.blue.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Kidney.blue.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Kidney.blue.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Kidney.blue.merged$Var1) + xlab("Modules")
print(p)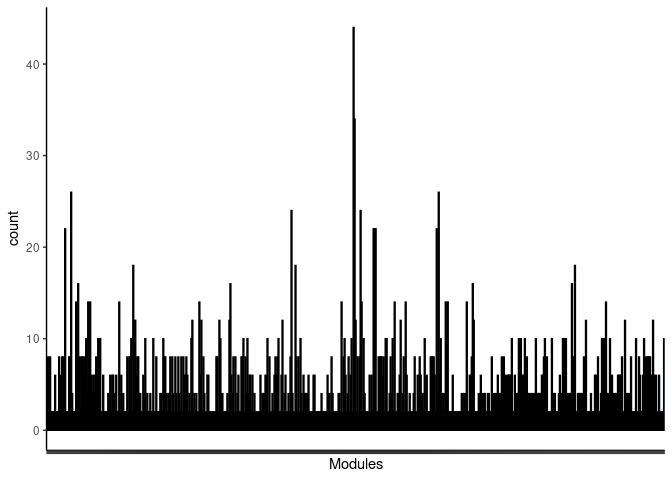
#dev.off()Pink
jaccard.Kidney.pink <- filtered.pathways[which(str_detect(names(filtered.pathways),"Kidney_pink"))
]
for(i in 1:length(jaccard.Kidney.pink)){
tissue <- str_split(names(jaccard.Kidney.pink), pattern = "_")[[i]][3]
kidney <- str_split(names(jaccard.Kidney.pink), pattern = "_")[[i]][1]
jaccard.Kidney.pink[[i]] <- jaccard.Kidney.pink[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(kidney == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Kidney.pink.merged <- map_df(jaccard.Kidney.pink, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Kidney.pink.merged <- jaccard.Kidney.pink.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Kidney.pink.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Kidney.pink.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Kidney.pink.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Kidney.pink.merged$Var1) + xlab("Modules")
print(p)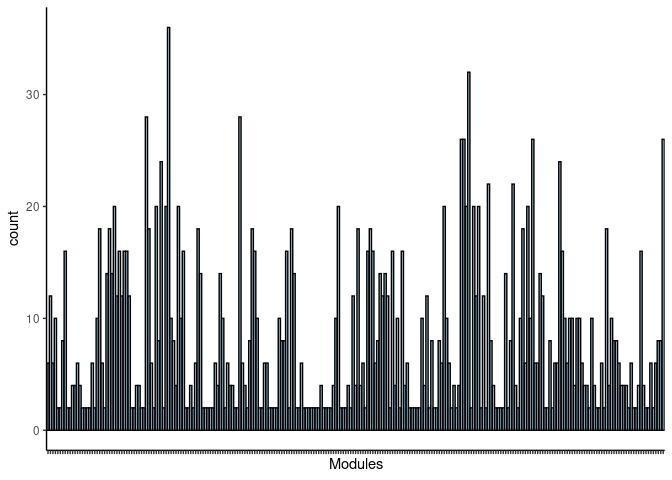
#dev.off()Liver
Darkturquoise
jaccard.Liver.darkturquoise <- filtered.pathways[which(str_detect(names(filtered.pathways),"Liver_darkturquoise"))
]
for(i in 1:length(jaccard.Liver.darkturquoise)){
tissue <- str_split(names(jaccard.Liver.darkturquoise), pattern = "_")[[i]][3]
Liver <- str_split(names(jaccard.Liver.darkturquoise), pattern = "_")[[i]][1]
jaccard.Liver.darkturquoise[[i]] <- jaccard.Liver.darkturquoise[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(Liver == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Liver.darkturquoise.merged <- map_df(jaccard.Liver.darkturquoise, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Liver.darkturquoise.merged <- jaccard.Liver.darkturquoise.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Liver.darkturquoise.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Liver.darkturquoise.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Liver.darkturquoise.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Liver.darkturquoise.merged$Var1) + xlab("Modules")
print(p)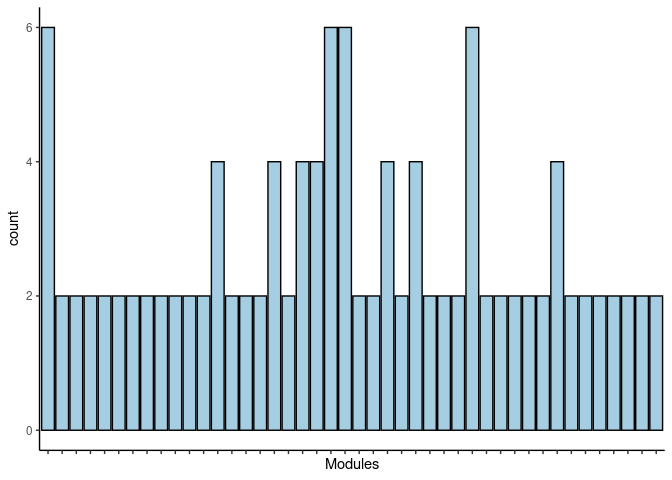
#dev.off()Darkolivegreen
jaccard.Liver.darkolivegreen <- filtered.pathways[which(str_detect(names(filtered.pathways),"Liver_darkolivegreen"))
]
for(i in 1:length(jaccard.Liver.darkolivegreen)){
tissue <- str_split(names(jaccard.Liver.darkolivegreen), pattern = "_")[[i]][3]
Liver <- str_split(names(jaccard.Liver.darkolivegreen), pattern = "_")[[i]][1]
jaccard.Liver.darkolivegreen[[i]] <- jaccard.Liver.darkolivegreen[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(Liver == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Liver.darkolivegreen.merged <- map_df(jaccard.Liver.darkolivegreen, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Liver.darkolivegreen.merged <- jaccard.Liver.darkolivegreen.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Liver.darkolivegreen.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Liver.darkolivegreen.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Liver.darkolivegreen.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Liver.darkolivegreen.merged$Var1) + xlab("Modules")
print(p)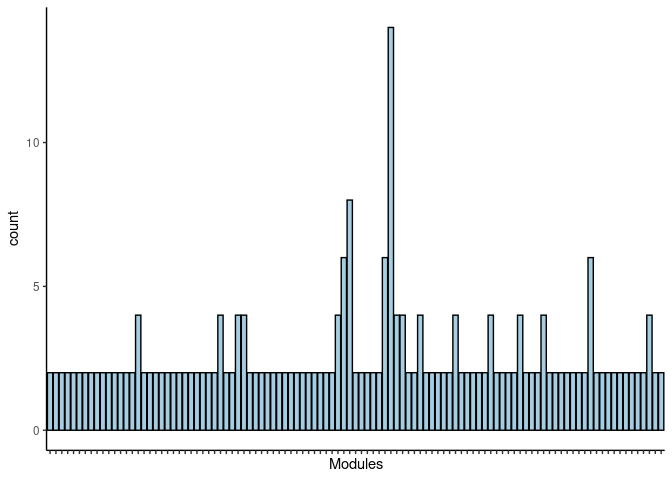
#dev.off()Skyblue3
jaccard.Liver.skyblue3 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Liver_skyblue3"))
]
for(i in 1:length(jaccard.Liver.skyblue3)){
tissue <- str_split(names(jaccard.Liver.skyblue3), pattern = "_")[[i]][3]
Liver <- str_split(names(jaccard.Liver.skyblue3), pattern = "_")[[i]][1]
jaccard.Liver.skyblue3[[i]] <- jaccard.Liver.skyblue3[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(Liver == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Liver.skyblue3.merged <- map_df(jaccard.Liver.skyblue3, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Liver.skyblue3.merged <- jaccard.Liver.skyblue3.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Liver.skyblue3.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Liver.skyblue3.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Liver.skyblue3.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Liver.skyblue3.merged$Var1) + xlab("Modules")
print(p)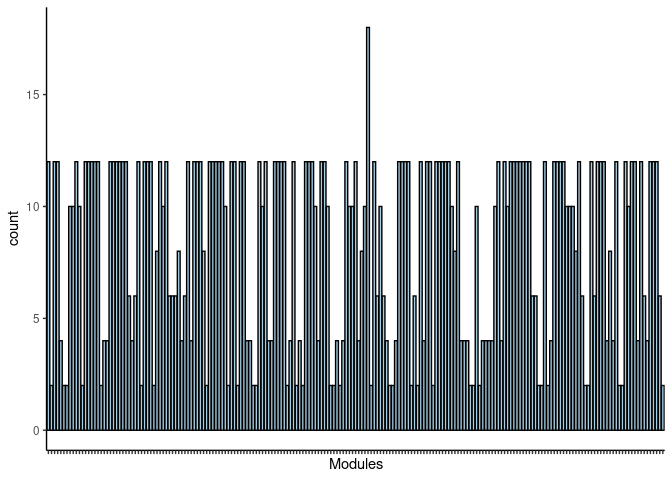
#dev.off()Prefrontal Cortex
Limegreen
jaccard.Prefrontal.Cortex.limegreen <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_limegreen"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.limegreen)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.limegreen), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.limegreen), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.limegreen[[i]] <- jaccard.Prefrontal.Cortex.limegreen[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.limegreen.merged <- map_df(jaccard.Prefrontal.Cortex.limegreen, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.limegreen.merged <- jaccard.Prefrontal.Cortex.limegreen.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.limegreen.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.limegreen.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.limegreen.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.limegreen.merged$Var1) + xlab("Modules")
print(p)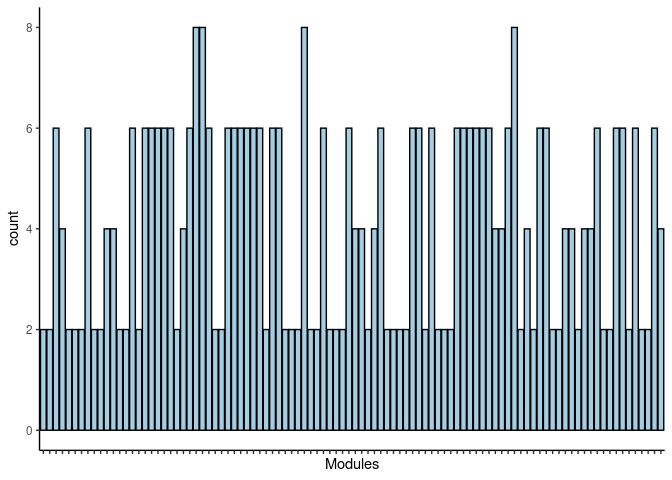
#dev.off()Rosybrown2
jaccard.Prefrontal.Cortex.rosybrown2 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_rosybrown2"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.rosybrown2)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.rosybrown2), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.rosybrown2), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.rosybrown2[[i]] <- jaccard.Prefrontal.Cortex.rosybrown2[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.rosybrown2.merged <- map_df(jaccard.Prefrontal.Cortex.rosybrown2, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.rosybrown2.merged <- jaccard.Prefrontal.Cortex.rosybrown2.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.rosybrown2.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.rosybrown2.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.rosybrown2.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.rosybrown2.merged$Var1) + xlab("Modules")
print(p)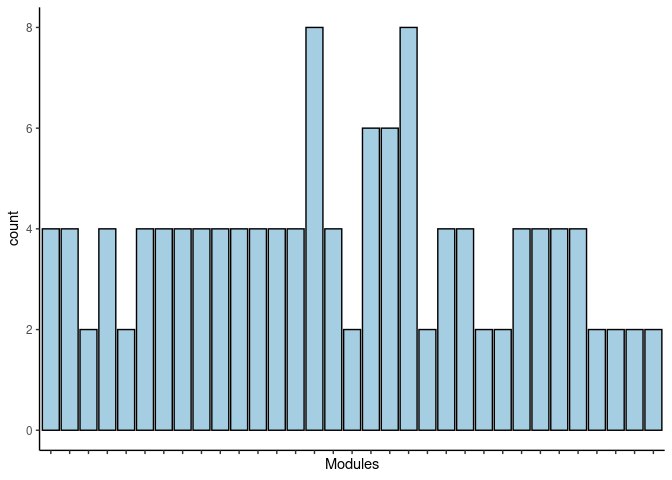
#dev.off()Lavenderblush2
jaccard.Prefrontal.Cortex.lavenderblush2 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_lavenderblush2"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.lavenderblush2)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.lavenderblush2), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.lavenderblush2), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.lavenderblush2[[i]] <- jaccard.Prefrontal.Cortex.lavenderblush2[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.lavenderblush2.merged <- map_df(jaccard.Prefrontal.Cortex.lavenderblush2, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.lavenderblush2.merged <- jaccard.Prefrontal.Cortex.lavenderblush2.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.lavenderblush2.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.lavenderblush2.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.lavenderblush2.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.lavenderblush2.merged$Var1) + xlab("Modules")
print(p)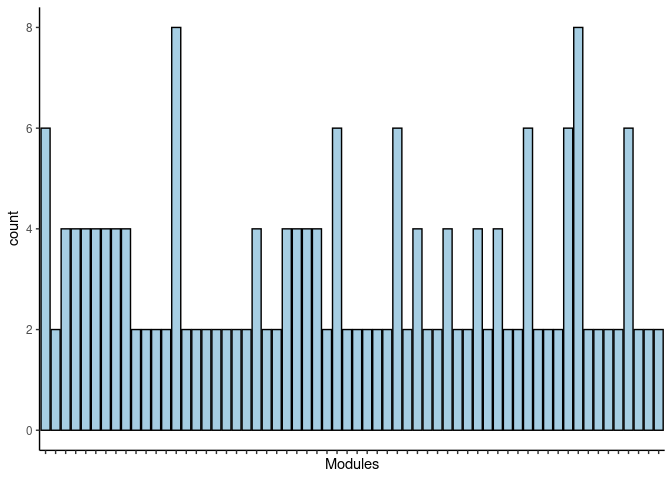
#dev.off()Antiquewhite
jaccard.Prefrontal.Cortex.antiquewhite <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_antiquewhite"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.antiquewhite)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.antiquewhite), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.antiquewhite), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.antiquewhite[[i]] <- jaccard.Prefrontal.Cortex.antiquewhite[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.antiquewhite.merged <- map_df(jaccard.Prefrontal.Cortex.antiquewhite, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.antiquewhite.merged <- jaccard.Prefrontal.Cortex.antiquewhite.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.antiquewhite.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.antiquewhite.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.antiquewhite.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.antiquewhite.merged$Var1) + xlab("Modules")
print(p)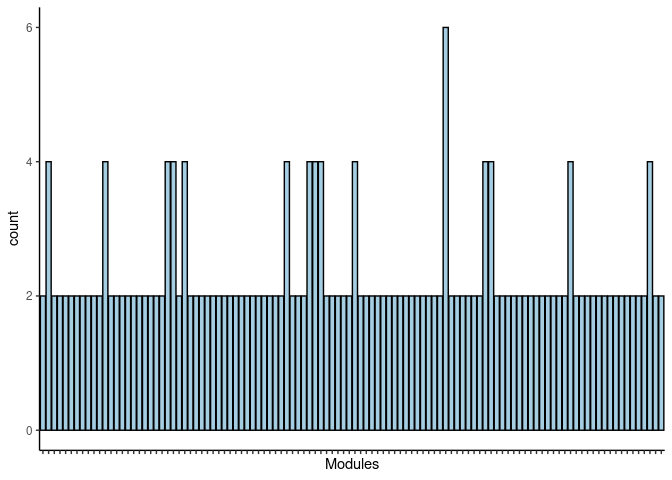
#dev.off()Pink
jaccard.Prefrontal.Cortex.pink <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_pink"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.pink)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.pink), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.pink), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.pink[[i]] <- jaccard.Prefrontal.Cortex.pink[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.pink.merged <- map_df(jaccard.Prefrontal.Cortex.pink, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.pink.merged <- jaccard.Prefrontal.Cortex.pink.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.pink.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.pink.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.pink.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.pink.merged$Var1) + xlab("Modules")
print(p)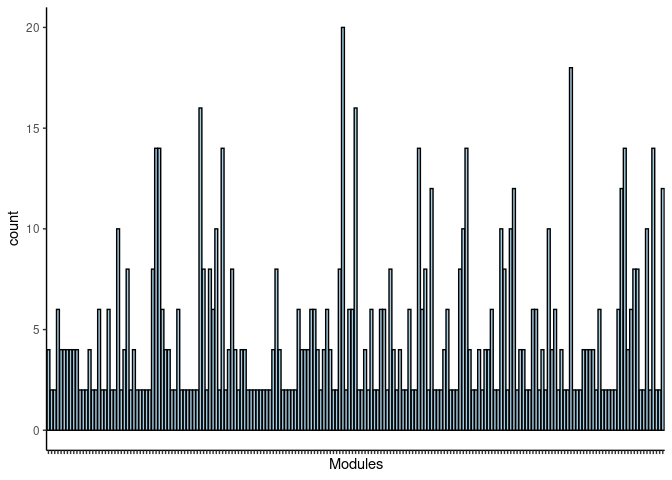
#dev.off()####Lightpink1
jaccard.Prefrontal.Cortex.lightpink1 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_lightpink1"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.lightpink1)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.lightpink1), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.lightpink1), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.lightpink1[[i]] <- jaccard.Prefrontal.Cortex.lightpink1[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.lightpink1.merged <- map_df(jaccard.Prefrontal.Cortex.lightpink1, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.lightpink1.merged <- jaccard.Prefrontal.Cortex.lightpink1.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.lightpink1.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.lightpink1.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.lightpink1.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.lightpink1.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Goldenrod3
jaccard.Prefrontal.Cortex.goldenrod3 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_goldenrod3"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.goldenrod3)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.goldenrod3), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.goldenrod3), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.goldenrod3[[i]] <- jaccard.Prefrontal.Cortex.goldenrod3[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.goldenrod3.merged <- map_df(jaccard.Prefrontal.Cortex.goldenrod3, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.goldenrod3.merged <- jaccard.Prefrontal.Cortex.goldenrod3.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.goldenrod3.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.goldenrod3.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.goldenrod3.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.goldenrod3.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Aquamarine
jaccard.Prefrontal.Cortex.aquamarine <- filtered.pathways[which(str_detect(names(filtered.pathways),"Prefrontal Cortex_aquamarine"))
]
for(i in 1:length(jaccard.Prefrontal.Cortex.aquamarine)){
tissue <- str_split(names(jaccard.Prefrontal.Cortex.aquamarine), pattern = "_")[[i]][3]
prefrontal <- str_split(names(jaccard.Prefrontal.Cortex.aquamarine), pattern = "_")[[i]][1]
jaccard.Prefrontal.Cortex.aquamarine[[i]] <- jaccard.Prefrontal.Cortex.aquamarine[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(prefrontal == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Prefrontal.Cortex.aquamarine.merged <- map_df(jaccard.Prefrontal.Cortex.aquamarine, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Prefrontal.Cortex.aquamarine.merged <- jaccard.Prefrontal.Cortex.aquamarine.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Prefrontal.Cortex.aquamarine.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Prefrontal.Cortex.aquamarine.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Prefrontal.Cortex.aquamarine.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Prefrontal.Cortex.aquamarine.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Skeletal Muscle
Grey
jaccard.Skeletal.Muscle.grey <- filtered.pathways[which(str_detect(names(filtered.pathways),"Skeletal Muscle_grey"))
]
for(i in 1:length(jaccard.Skeletal.Muscle.grey)){
tissue <- str_split(names(jaccard.Skeletal.Muscle.grey), pattern = "_")[[i]][3]
muscle <- str_split(names(jaccard.Skeletal.Muscle.grey), pattern = "_")[[i]][1]
jaccard.Skeletal.Muscle.grey[[i]] <- jaccard.Skeletal.Muscle.grey[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue== "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(muscle == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(tissue == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Skeletal.Muscle.grey.merged <- map_df(jaccard.Skeletal.Muscle.grey, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Skeletal.Muscle.grey.merged <- jaccard.Skeletal.Muscle.grey.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Skeletal.Muscle.grey.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Skeletal.Muscle.grey.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Skeletal.Muscle.grey.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Skeletal.Muscle.grey.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Small Intestine
Green4
jaccard.Small.Intestine.green4 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Small Intestine_green4"))
]
for(i in 1:length(jaccard.Small.Intestine.green4)){
tissue <- str_split(names(jaccard.Small.Intestine.green4), pattern = "_")[[i]][3]
intestine <- str_split(names(jaccard.Small.Intestine.green4), pattern = "_")[[i]][1]
jaccard.Small.Intestine.green4[[i]] <- jaccard.Small.Intestine.green4[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(intestine == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Small.Intestine.green4.merged <- map_df(jaccard.Small.Intestine.green4, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Small.Intestine.green4.merged <- jaccard.Small.Intestine.green4.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Small.Intestine.green4.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Small.Intestine.green4.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Small.Intestine.green4.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Small.Intestine.green4.merged$Var1) + xlab("Modules")
print(p)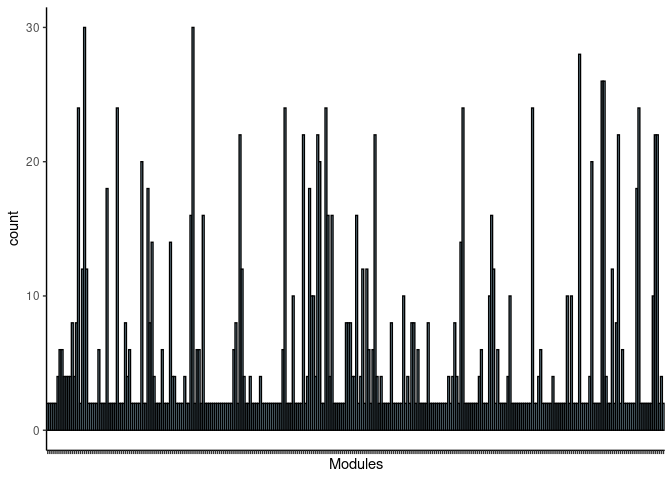
#dev.off()Blue2
jaccard.Small.Intestine.blue2 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Small Intestine_blue2"))
]
for(i in 1:length(jaccard.Small.Intestine.blue2)){
tissue <- str_split(names(jaccard.Small.Intestine.blue2), pattern = "_")[[i]][3]
intestine <- str_split(names(jaccard.Small.Intestine.blue2), pattern = "_")[[i]][1]
jaccard.Small.Intestine.blue2[[i]] <- jaccard.Small.Intestine.blue2[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(intestine == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Small.Intestine.blue2.merged <- map_df(jaccard.Small.Intestine.blue2, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Small.Intestine.blue2.merged <- jaccard.Small.Intestine.blue2.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Small.Intestine.blue2.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Small.Intestine.blue2.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Small.Intestine.blue2.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Small.Intestine.blue2.merged$Var1) + xlab("Modules")
print(p)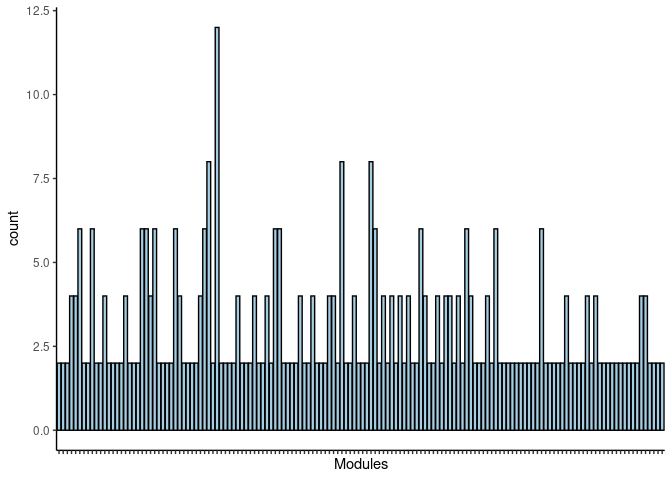
#dev.off()Cornsilk
jaccard.Small.Intestine.cornsilk <- filtered.pathways[which(str_detect(names(filtered.pathways),"Small Intestine_cornsilk"))
]
for(i in 1:length(jaccard.Small.Intestine.cornsilk)){
tissue <- str_split(names(jaccard.Small.Intestine.cornsilk), pattern = "_")[[i]][3]
intestine <- str_split(names(jaccard.Small.Intestine.cornsilk), pattern = "_")[[i]][1]
jaccard.Small.Intestine.cornsilk[[i]] <- jaccard.Small.Intestine.cornsilk[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(intestine == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Small.Intestine.cornsilk.merged <- map_df(jaccard.Small.Intestine.cornsilk, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Small.Intestine.cornsilk.merged <- jaccard.Small.Intestine.cornsilk.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Small.Intestine.cornsilk.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Small.Intestine.cornsilk.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Small.Intestine.cornsilk.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Small.Intestine.cornsilk.merged$Var1) + xlab("Modules")
print(p)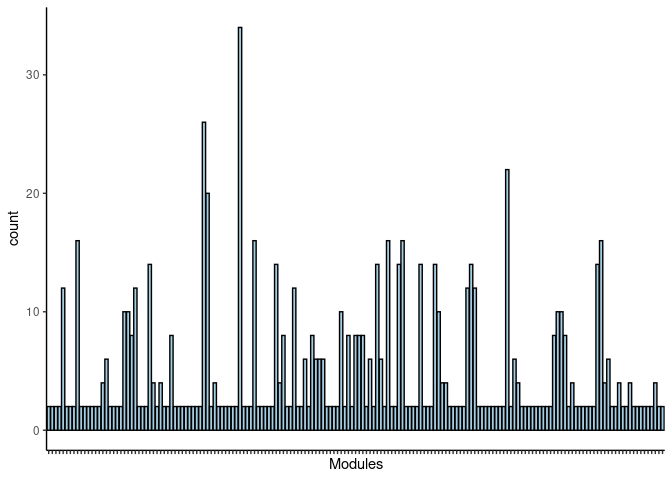
#dev.off()Magenta2
jaccard.Small.Intestine.magenta2 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Small Intestine_magenta2"))
]
for(i in 1:length(jaccard.Small.Intestine.magenta2)){
tissue <- str_split(names(jaccard.Small.Intestine.magenta2), pattern = "_")[[i]][3]
intestine <- str_split(names(jaccard.Small.Intestine.magenta2), pattern = "_")[[i]][1]
jaccard.Small.Intestine.magenta2[[i]] <- jaccard.Small.Intestine.magenta2[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(intestine == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Small.Intestine.magenta2.merged <- map_df(jaccard.Small.Intestine.magenta2, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Small.Intestine.magenta2.merged <- jaccard.Small.Intestine.magenta2.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Small.Intestine.magenta2.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Small.Intestine.magenta2.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Small.Intestine.magenta2.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Small.Intestine.magenta2.merged$Var1) + xlab("Modules")
print(p)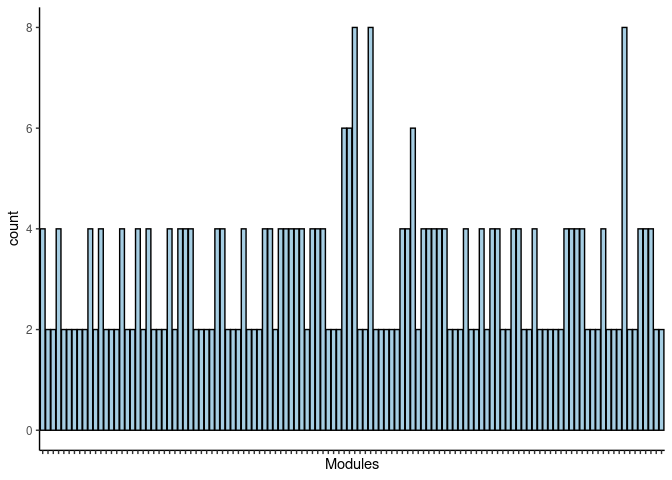
#dev.off()Darkgoldenrod4
jaccard.Small.Intestine.darkgoldenrod4 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Small Intestine_darkgoldenrod4"))
]
for(i in 1:length(jaccard.Small.Intestine.darkgoldenrod4)){
tissue <- str_split(names(jaccard.Small.Intestine.darkgoldenrod4), pattern = "_")[[i]][3]
intestine <- str_split(names(jaccard.Small.Intestine.darkgoldenrod4), pattern = "_")[[i]][1]
jaccard.Small.Intestine.darkgoldenrod4[[i]] <- jaccard.Small.Intestine.darkgoldenrod4[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(intestine == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Small.Intestine.darkgoldenrod4.merged <- map_df(jaccard.Small.Intestine.darkgoldenrod4, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Small.Intestine.darkgoldenrod4.merged <- jaccard.Small.Intestine.darkgoldenrod4.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Small.Intestine.darkgoldenrod4.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Small.Intestine.darkgoldenrod4.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Small.Intestine.darkgoldenrod4.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Small.Intestine.darkgoldenrod4.merged$Var1) + xlab("Modules")
print(p)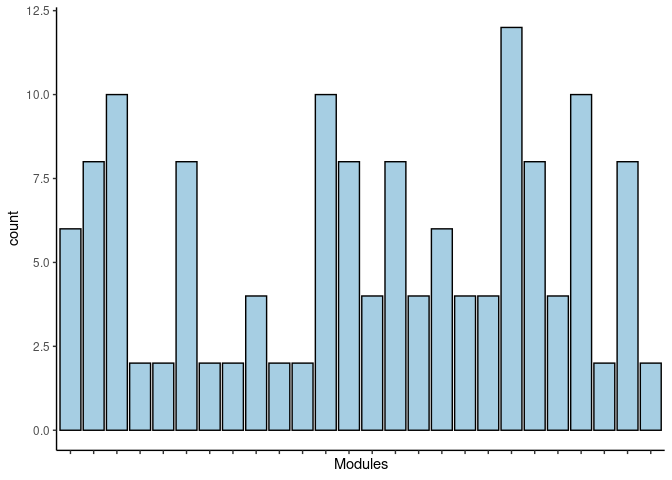
#dev.off()Spleen
Tan4
jaccard.Spleen.tan4 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Spleen_tan4"))
]
for(i in 1:length(jaccard.Spleen.tan4)){
tissue <- str_split(names(jaccard.Spleen.tan4), pattern = "_")[[i]][3]
spleen <- str_split(names(jaccard.Spleen.tan4), pattern = "_")[[i]][1]
jaccard.Spleen.tan4[[i]] <- jaccard.Spleen.tan4[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(spleen == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Spleen.tan4.merged <- map_df(jaccard.Spleen.tan4, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Spleen.tan4.merged <- jaccard.Spleen.tan4.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Spleen.tan4.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Spleen.tan4.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Spleen.tan4.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Spleen.tan4.merged$Var1) + xlab("Modules")
print(p)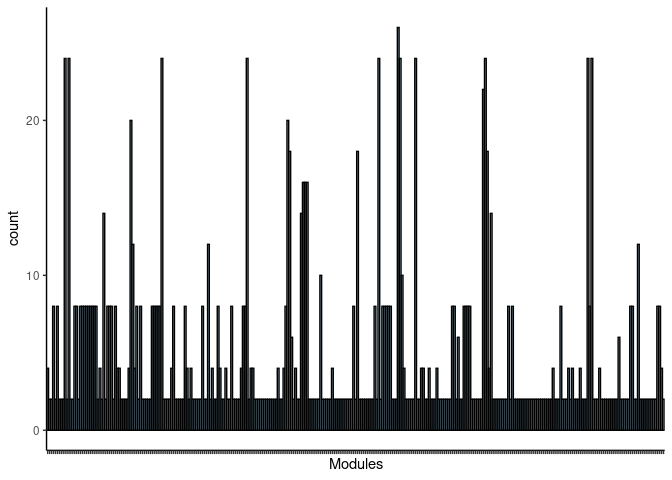
#dev.off()Mediumpurple1
jaccard.Spleen.mediumpurple1 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Spleen_mediumpurple1"))
]
for(i in 1:length(jaccard.Spleen.mediumpurple1)){
tissue <- str_split(names(jaccard.Spleen.mediumpurple1), pattern = "_")[[i]][3]
spleen <- str_split(names(jaccard.Spleen.mediumpurple1), pattern = "_")[[i]][1]
jaccard.Spleen.mediumpurple1[[i]] <- jaccard.Spleen.mediumpurple1[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(spleen == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Spleen.mediumpurple1.merged <- map_df(jaccard.Spleen.mediumpurple1, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Spleen.mediumpurple1.merged <- jaccard.Spleen.mediumpurple1.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Spleen.mediumpurple1.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Spleen.mediumpurple1.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Spleen.mediumpurple1.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Spleen.mediumpurple1.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Palevioletred3
jaccard.Spleen.palevioletred3 <- filtered.pathways[which(str_detect(names(filtered.pathways),"Spleen_palevioletred3"))
]
for(i in 1:length(jaccard.Spleen.palevioletred3)){
tissue <- str_split(names(jaccard.Spleen.palevioletred3), pattern = "_")[[i]][3]
spleen <- str_split(names(jaccard.Spleen.palevioletred3), pattern = "_")[[i]][1]
jaccard.Spleen.palevioletred3[[i]] <- jaccard.Spleen.palevioletred3[[i]] %>%
mutate(Heart = if(tissue == "Heart") {"yes"} else {"no"}) %>%
mutate(Hippocampus = if(tissue == "Hippocampus") {"yes"} else {"no"}) %>%
mutate(Hypothalamus = if(tissue == "Hypothalamus") {"yes"} else {"no"}) %>%
mutate(Kidney = if(tissue == "Kidney") {"yes"} else {"no"}) %>%
mutate(Liver = if(tissue == "Liver") {"yes"} else {"no"}) %>%
mutate("Prefrontal_Cortex" = if(tissue == "Prefrontal Cortex") {"yes"} else {"no"}) %>%
mutate("Skeletal_Muscle" = if(tissue == "Skeletal Muscle") {"yes"} else {"no"}) %>%
mutate("Small_Intestine" = if(spleen == "Small Intestine") {"yes"} else {"no"}) %>%
mutate(Spleen = if(tissue == "Spleen") {"yes"} else {"no"})
}
jaccard.Spleen.palevioletred3.merged <- map_df(jaccard.Spleen.palevioletred3, ~as.data.frame(.)) %>% group_by(term_name, Heart, Hippocampus, Hypothalamus, Kidney, Liver, Prefrontal_Cortex, Skeletal_Muscle, Small_Intestine, Spleen) %>% summarise(count = n())
jaccard.Spleen.palevioletred3.merged <- jaccard.Spleen.palevioletred3.merged %>% group_by(term_name) %>% summarise_each(funs(max)) Frequency Table
DT.table.jaccard.pathways(jaccard.Spleen.palevioletred3.merged)Frequency Plot
#pdf("Counts_of_each_pathway_identified_within_jaccard_index.pdf")
p <- ggplot(data=jaccard.Spleen.palevioletred3.merged,aes(x=term_name,y=count))
p <- p + geom_bar(color="black", fill=colorRampPalette(brewer.pal(n = 12, name = "Paired"))(length(jaccard.Spleen.palevioletred3.merged[,1])), stat="identity",position="identity") + theme_classic()
p <- p + theme(axis.text.x = element_text(angle = 90, hjust =1, size = 3)) + scale_x_discrete(labels=jaccard.Spleen.palevioletred3.merged$Var1) + xlab("Modules")
print(p)
#dev.off()Analysis performed by Ann Wells
The Carter Lab The Jackson Laboratory 2023
ann.wells@jax.org